
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Опухолевые супрессоры и мутаторные гены
Российский онкологический научный центр им. РАМН, Москва
Введение
Ключевую роль в возникновении важнейших свойств неопластической клетки играют нарушения функции протоонкогенов, опухолевых супрессоров и так называемых мутаторных генов. Термином опухолевые супрессоры (другие названия - антионкогены, рецессивные опухолевые гены) принято обозначать гены, инактивация функции которых ведет к возникновению и/или прогрессии новообразований. И, наоборот, восстановление экспрессии некоторых из таких генов в опухолевых клетках может подавить их дальнейшее размножение. Мутаторными называют гены, нарушения функции которых тем или иным способом увеличивает темп возникновения мутаций и/или других генетических изменений. Следует заметить, что многие из опухолевых супрессоров являются одновременно и мутаторными генами. Инактивация таких генов столь сильно увеличивает вероятность появления различных онкогенных мутаций, что образование опухоли становится лишь делом времени. Действительно, врожденные мутации даже в одном из аллелей некоторых из опухолевых супрессоров/мутаторных генов делает очень вероятным возникновение в молодом возрасте определенных форм новообразований: сарком, лейкозов, опухолей мозга или раков молочной железы (синдром Ли-Фраумени) - при мутациях р53; раков молочной железы и яичников - при мутациях генов BRCA1 и BRCA2; и т. д. (см. Табл. 1).
Табл.1. Основные характеристики некоторых идентифицированных опухолевых супрессоров и мутаторных генов
|
*Подчеркнуты наследственные формы заболеваний, возникающие при мутациях в половых клетках.
** Ген INK4a кодирует два белка: p16INK4a и p14ARF (Alternative Reading Frame) - продукт альтернативной рамки считывания. Делеции и многие точечные мутации в гене INK4a вызывают одновременную инактивацию супрессорных активностей обоих этих белков.
Доказательством причинной роли таких мутаций в канцерогенезе являются результаты экспериментов по созданию линий трансгенных мышей, во всех клетках которых инактивированы ("нокаутированы") один или оба аллеля какого-либо из опухолевых супрессоров или мутаторных генов. Если такие изменения не вызывают внутриутробную гибель, то у рождающихся животных возникает сильная предрасположенность к развитию определенных новообразований. Так, мыши с инактивированным р53 характеризуются почти 100%-ной вероятностью развития в молодом возрасте новообразований, спектр которых сходен с наблюдаемым при синдроме Ли-Фраумени.
Геном человека содержит по меньшей мере несколько десятков опухолевых супрессоров и мутаторных генов. Более 30 из них уже идентифицированы, для многих известны выполняемые в клетке функции (см. Табл. 1). Кроме того, выявлено еще несколько десятков участков хромосом, потери которых обнаруживаются во многих случаях какой-то определенной формы опухолей или в различных новообразованиях, что указывает на возможную локализацию в них потенциальных опухолевых супрессоров. Ниже будут рассмотрены основные сведения о функциях известных опухолевых супрессоров/мутаторных генах и возможных механизмах возникновения новообразований при их нарушениях.
pRb - первый идентифицированный опухолевый супрессор
Идентификация опухолевых супрессоров началась с обнаружения гена Rb, врожденные мутации которого вызывают развитие ретинобластом. В начале 1970-х г. г. Кнудсон, проводя эпидемиологические исследования, отметил, что около 40% ретинобластом возникает в младенческом возрасте (средний возраст 14 месяцев). Причем эти опухоли, как правило, билатеральные (возникают из сетчатки обоих глаз) и часто множественные (в среднем по 3 независимых опухоли на пациента). Если такие пациенты в результате хирургического вмешательства излечивались от ретинобластом, у многих из них в юношеском возрасте развивались остеосаркомы, а в зрелом возрасте - меланомы кожи. При этом во многих случаях отмечался наследственный характер заболевания. Кнудсон сформулировал гипотезу, согласно которой, если дети наследуют мутантный аллель гена, позже названного Rb, то вторая мутация, происходящая уже в ретинобласте, ведет к возникновению опухоли. Это довольно частое событие (происходит примерно у 95% пациентов с частотой около трех на пациента) и, поэтому, предрасположение к развитию болезни имеет доминантный характер наследования. Кнудсон предположил также, что дети, у которых ретинобластомы возникают в более позднем возрасте (частота таких опухолей невелика - 1 на 30000 индивидуумов), не наследуют мутантный аллель гена Rb. Вместо этого у них происходят две независимые мутации в одном из ретинобластов, что и приводит к развитию опухоли. Следовательно, согласно гипотезе Кнудсона, у первой группы пациентов имеется одна врожденная и одна приобретенная мутации, тогда как у второй группы больных обе мутации приобретенные.
Так как при некоторых наследственных ретинобластомах обнаруживались небольшие делеции участка длинного плеча хромосомыq14), было предположено, что ген "предрасположенности к ретинобластоме" (Rb) локализуется именно в этом участке генома. И действительно, с помощью позиционного клонирования был изолирован ген, оба аллеля которого инактивированы в клетках как наследственных, так и спорадических ретинобластом. При этом при наследственных формах заболевания все клетки организма имели врожденные мутации этого гена. Таким образом, стало ясно, что постулируемые Кнудсоном две мутации, необходимые для развития ретинобластом, происходят в разных аллелях одного и того же гена Rb. Инактивация гена Rb была обнаружена не только в ретинобластомах, но и в клетках некоторых других, ненаследственных новообразований: практически во всех случаях мелкоклеточного рака легкого, части случаев (20-40%) острого лейкоза, остеосаркомы, рака мочевого пузыря, простаты и др. Восстановление экспрессии этого гена в культивируемых in vitro клетках ретинобластом и остеосарком приводило к торможению их роста. Таким образом, впервые были получены веские доказательства существования генов, полная инактивация которых приводит к развитию новообразований и продукты которых способны подавить размножение неопластических клеток. Закономерности, выявленные при исследовании гена Rb, в частности ассоциация с наследственными формами опухолей и необходимость поражения обоих аллелей (рецессивный характер проявления мутаций), стали использоваться в качестве критериев при поиске и идентификации других опухолевых супрессоров.
В настоящее время достигнут значительный прогресс в понимании нормальной функции продукта гена Rb (pRb) в клетке, охарактеризованы типы и местоположение мутаций в гене Rb при ретинобластомах и других новообразованиях, предложены высокоэффективные методы скрининга мутаций Rb у индивидуумов в группах риска.
Функция pRb в клетке и ее нарушения при канцерогенезе
pRb представляет собой фосфобелок c мол. массой 105кД, локализующийся в ядре и экспрессирующийся в большинстве типов клеток. Он дефосфорилирован в неделящихся клетках, а также в пролиферирующих клетках, находящихся в начале G1 фазы клеточного цикла. В таком состоянии pRb образует комплексы с рядом белков, в том числе с белками, вызывающими ремоделирование хроматина (гистоновые деацетилазы HDAC, комплексы SWI-SNF) и транскрипционными факторами семейства E2F, регулирующими активность генов, продукты которых необходимы для начала и прохождения S-фазы (циклин Е, циклин А, дигидрофолатредуктаза, тимидинкиназа, PCNA, ДНК-полимераза a и др). Транскрипция этих генов подавлена, если E2F связан с комплексами HDAC/pRb/SWI-SNF. При митогенных сигналах pRb в середине G1 фазы фосфорилируется по определенным аминокислотным остаткам циклинзависимой киназой циклин D/Сdk4(6), что вызывает отвязывание от него HDAC. Комплекс pRb/SWI-SN (без HDAC) не блокирует способность E2F транс-активировать ген циклина E, но репрессирует транскрипцию других E2F-регулируемых генов, в частности циклина А. В результате в конце G1 фазы происходит избирательная активация циклинзависимой киназы циклин Е/Cdk2, которая дополнительно фосфорилирует pRb по другим аминокислотным остаткам. Это вызывает высвобождение транскрипционного фактора E2F из комплексов с pRb/SWI-SNF и его активацию, приводящую к повышению экспрессии циклина А и других генов, продукты которых необходимы для синтеза ДНК (Рис. 1).
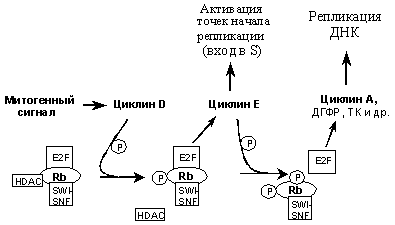
Рис. 1. pRb регулирует активность транскрипционных факторов семейства E2F и циклинзависимых киназ, ответственных за вход и продвижение по S фазе клеточного цикла (объяснения в тексте).
После завершения S фазы pRb переходит в дефосфорилированное состояние, в котором он блокирует активность E2F и вход в следующую S фазу (для ее инициации необходим новый митогенный стимул, активирующий комплексы циклин D/Cdk4). Таким образом, модулируя активность E2F и регулируемых им генов, pRb играет ключевую роль в контроле последовательности событий, обеспечивающих переход клетки из G0/G1 в S фазу и ее успешное завершение.
В дополнение к E2F pRb связывает и модулирует активность ряда других транскрипционных факторов, большинство из которых тканеспецифичны и участвуют в регуляции определенных дифференцировочных программ. Так, он увеличивает транскрипционную активность некоторых представителей семейства bHLH (MyoD и др.), которые стимулируют мышечную дифференцировку и семейства C/EBP, играющих ключевую роль в моноцитарно-макрофагальной (NF-IL6) и адипоцитарной (C/EBP-b) дифференцировках. Кроме того, pRb связывает и репрессирует гомеобокс-содержащие транскрипционные факторы (Pax-3, Mhox, Chx10), определяющие судьбу клетки в раннем эмбрионенезе. Все это дало основание полагать, что основная физиологическая функция pRb заключается в детерминировании некоторых терминальных дифференцировок, для чего необходима остановка клеточного цикла (осуществляется за счет подавления функции E2F) и индукция экспрессии ряда специализированных белков (реализуется путем модификации активности тканеспецифичных транскрипционных факторов).
Значительная часть мутаций гена Rb, обнаруживаемых в различных опухолях, вызывают либо делецию гена, либо сдвиг кодирующей рамки, либо ее преждевременную терминацию, либо нарушения сплайсинга мРНК. Все это приводит или к полной потере экспрессии белкового продукта, или к экспрессии неполноценных и нестабильных белков pRb. В части случаев наблюдаются миссенс-мутации, вызывающие замену одного из аминокислотных остатков. Такие мутации поражают домен, ответственный за связывание pRb c рядом клеточных белков и вирусными онкобелками - T-SV40, E1A аденовирусов и E7 HPV. По-видимому нарушение функции именно этого домена, вызываемое либо мутациями, либо его связыванием с вирусными онкобелками, является критичным для подавления супрессорной активности pRb.
При мутациях обоих аллелей гена Rb, вызывающих отсутствие в клетке белка pRb или экспрессию его функционально неактивной формы, транскрипционный фактор E2F находится в перманентно активированном состоянии. Это, во-первых, уменьшает зависимость размножения клеток от ростовых факторов, а во-вторых, отменяет негативную регуляцию клеточной пролиферации при рост-ингибирующих сигналах. Кроме того, потеря функции pRb нарушает процессы клеточной дифференцировки. Все это резко увеличивают вероятность появления постоянно пролиферирующих клонов клеток, в которых будут накапливаться и другие онкогенные мутации, ведущие к злокачественной трансформации. При этом, пока остается неясным, почему при врожденной инактивации одного из аллелей гена Rb во всех клетках организма у пациента в юном возрасте развивается именно ретинобластома, а не какие-то другие новообразования. Интересно, что у трансгенных мышей с инактивацией одного из аллелей гена Rb (инактивация обоих аллелей несовместима с жизнью эмбрионов из-за нарушений эритропоэза и нейрогенеза) возникают не ретинобластомы, а медуллярные раки щитовидной железы или аденомы средней доли гипофиза, возникающие из меланофоров (в отличие от мышей у человека эта ткань в гипофизе атрофирована). Более того, ретинобластомы не развиваются и у химерных мышей, во всех клетках сетчатки которых инактивированы оба аллеля гена Rb. Отсутствие ретинобластом у таких химерных мышей может быть объяснено относительно небольшим, по сравнению с человеком, количеством клеток в сетчатке, что уменьшает вероятность возникновения в течение короткой жизни животного в одном из ретинобластов дополнительных генетических изменений, необходимых для развития новообразования. Меньшая вероятность таких событий может быть также связана с отсутствием у лабораторных мышей дополнительных канцерогенных факторов, таких как облучение клеток сетчатки солнечным светом, способствующему, очевидно, развитию ретинобластом в аналогичной ситуации у человека. В пользу предположения о необходимости для развития ретинобластом дополнитльных событий может свидетельствовать тот факт, что ретинобластомы возникают у трансгенных мышей, экспрессирующих вирусный онкобелок T-SV40, который связывает и инактивирует не только pRb, но и его гомологи (см. ниже), а также другой опухолевый супрессор - р53 (см. раздел 3.3).
Гомологи pRb: p107 и Rb2/p130
Несколько позже, в начале 90-х гг. было идентифициоровано два гена, продукты которых, белки р107 и Rb2/p130, имеют структурное сходство и частично перекрывающиеся функции с pRb. Так, подобно pRb, они способны подавлять активность E2F-респонсивных генов и блокировать вход в фазу S. Вместе с тем, они имеют ряд отличий от pRb. В частности, они связывают только E2F4 и E2F5, тогда как рRb взаимодействует с E2F1, E2F2, E2F3 и E2F4. Возможно поэтому р107 и Rb2/p130, в отличии от pRb, не способны поддерживать длительное пребывание в G0 и дифференцировку некоторых типов клеток, например миоцитов.
Нарушения функции гомологов pRb, по-видимому, не причастны к инициальным этапам развития каких-либо опухолей человека, так как пока не выявлены наследственные формы новообразований с врожденными герминальными мутациями генов р107 или Rb2/p130. Кроме того, у мышей с гетерозиготным нокаутом этих генов частота возникновения опухолей не повышается. В то же время соматические инактивирующие мутации гена Rb2/p130 характерны для части случаев лимфомы Беркита, рака носоглотки и мелкоклеточного рака легкого, причем, как правило, они выявляются на поздних стадиях заболевания. Вероятно, нарушения функции этого гомолога pRb обеспечивают прогрессию некоторых новообразований.
р53 - многофункциональный опухолевый супрессор, чаще всего поражаемый в различных новообразованиях человека
Типы опухолей, ассоциированные с аномалиями р53
Наиболее универсальным молекулярным изменением в различных новообразованиях человека является инактивация функции белка р53. Более чем в половине всех опухолей человека (50-60% новообразований более чем 50 различных типов) обнаруживаются мутации гена р53. В отличие от других опухолевых супрессоров, для которых характерны мутации, прекращающие синтез белка (делеции, образование стоп-кодонов, сдвиг кодирующей рамки, нарушения сплайсинга мРНК), подавляющее большинство (более 90%) мутаций р53 представляет собой миссенс-мутации, приводящие к замене одной из аминокислот в белковой молекуле на другую. Еще одной особенностью мутаций р53 в опухолевых клетках является то, что они, в отличие от мутаций других опухолевых супрессоров, часто являются гетерозиготными, т. е. поражают только один из двух аллелей гена. (Причины этих различий будут раскрыты ниже, при рассмотрении структурной организации и функций р53 - см. раздел 3.3.2.) Мутации обнаруживаются в разных участках молекулы р53, но чаще всего в его эволюционно консервативном ДНК-связывающем домене, причем с наибольшей частотой в кодонах 175, 245, 248, 249, 273 и 282 (так называемые горячие точки) - см. Рис. 2. Интересно, что спектр мутаций несколько меняется в зависимости от гистогенеза опухоли и/или этиологического фактора. Например, мутации в кодоне 175 не встречаются в опухолях легких, а замены в другой горячей точке - кодоне 273 - не обнаруживаются при бластном кризе хронического миелоидного лейкоза. В то же время для раков легкого характерны мутации в кодоне 145, очень редко встречающиеся в других новообразованиях, а мутации в кодоне 249 обнаруживаются преимущественно в гепатокарциномах, вызванных специфическим канцерогеном - афлатоксином В. Очевидно, отражением действия канцерогенов с разными механизмами мутагенного действия являются и отличия в характере мутаций р53 в разных опухолях. Так, при опухолях лёгких, печени и лимфомах замена аминокислотных остатков в большинстве случаев обусловлена трансверсиями (в ДНК пуриновый нуклеотид заменен на пиримидиновый или наоборот), а при карциномах кожи, лимфоме Беркитта, Т-клеточном лейкозе - транзициями (заменой пуринового основания на другое пуриновое или пиримидинового на другое пиримидиновое). Герминальные (произошедшие в половой клетке и передающиеся по наследству) мутации в одном из аллелей гена р53 вызывают синдром Ли-Фраумени, заключающийся во врожденном предрасположении к развитию различных новообразований, в первую очередь сарком, рака молочной железы, лимфолейкозов. Нередко синдром Ли-Фраумени характеризуется возникновением первично-множественных опухолей. Примечательно, что у трансгенных мышей, несущих инактивирующие мутации в гене р53, наблюдается картина, очень напоминающая синдром Ли-Фраумени. Примерно у трети животных, у которых инактивирован один из двух аллелей р53, в течение 6-9 месяцев после рождения возникают новообразования, причем их спектр очень сходен с наблюдаемым при синдроме Ли-Фраумени. При этом в части этих опухолей, как и новообразований у пациентов с синдромом Ли-Фраумени, сохраняется экспрессия неповрежденного аллеля гена р53. При врожденной инактивации во всех клетках организма обоих аллелей гена р53 опухоли развиваются практически у всех животных. Примерно такая же картина наблюдается у трансгенных мышей, несущих дополнительный экзогенный аллель р53, кодирующий белок с миссенс-мутацией.
Важно подчеркнуть, что мутации - не единственный путь нарушения функции белка р53 в опухолевых клетках. Так, для 10-20% рака молочной железы, а также для нейробластом характерно нарушение транспорта р53 из цитоплазмы в ядро, где он проявляет свою функциональную активность. В части остеосарком наблюдается амплификация клеточного онкогена MDM2, продукт которого связывает и инактивирует белок р53 (см. раздел 3.3.3). При раке шейки матки, ассоциированном с вирусами папиллом человека, происходит связывание р53 с вирусным онкобелком Е6), что вызывает деградацию белка р53, и т. д.
Структурная организация и биохимические активности белка р53
Продукт гена р53 имеет мол. массу 53кДа и состоит из 392 аминокислотных остатков. Он образует тетрамерный комплекс, способный регулировать транскрипцию ряда генов, имеющих в своем составе специфические последовательности ДНК, так называемые р53-респонсивные элементы. В молекуле р53 картировано несколько функционально-значимых доменов, играющих важную роль в осуществлении или регуляции его активности (Рис. 2). N-концевой участок (аминокислоты 1-42) представляет собой домен, ответственный за транскрипционную активацию генов-мишеней. Он обладает способностью связываться с компонентами базальных факторов транскрипции, в частности с субъединицами hTAFII31, hTAF70 комплекса TFIID РНК-полимеразы II, а также с транскрипционным кофактором p300/CBP. Кроме того, этот домен участвует в белок-белковых взаимодействиях, регулирующих стабильность молекулы р53. И, наконец, в нем расположено несколько остатков серина и треонина, фосфорилирование которых регулирует активность р53.
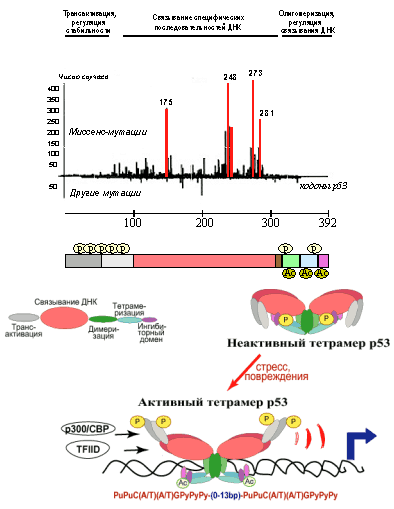
Рис. 2. Схематическое изображение функциональных доменов р53, предполагаемой модели приобретения белком транскрипционно активной конформации и частоты встречаемости в новообразованиях человека мутаций в разных участках молекулы р53.
Центральный домен р53 (аминокислоты 120-290) непосредственно узнает и связывает специфические последовательности ДНК регулируемых генов, так называемые р53-респонсивные элементы, состоящие из расположенных друг за другом последовательностей с обобщенной структурой типа PuPuC(A/T)(A/T)GPyPyPy (Pu - пурин, Py - пиримидин). Именно в этом ДНК-связывающем домене локализуется большинство точечных мутаций, обнаруживаемых в различных опухолях человека (Рис. 2). Далее идут участки ответственные за ядерную локализацию (аминокислоты 305-323) и димеризацию/тетрамеризацию молекул р53 (аминокислоты 323-356). С-концевой участок р53 (аминокислоты 363-392) представляет собой так называемый ингибиторный домен. В немодифицированном состоянии он препятствует посадке ДНК-связывающего домена на специфическую последовательность регулируемого гена. Фосфорилирование и ацетилирование его определенных сайтов вызывают изменения конформации белковой молекулы и переход тетрамеров р53 из неактивного (латентного) состояния в активное. В результате ДНК-связывающие домены освобождаются от блокирующего влияния ингибиторных доменов и приобретают способность садиться на р53-респонсивные элементы. Таким образом, к респонсивным генам привлекаются базовые факторы транскрипции, связывающиеся с N-концевым участком р53, и стимулируется синтез РНК генов-мишеней.
Помимо повышения транскрипции генов, содержащих специфические респонсивные элементы, белок р53 обладает также и рядом других активностей. В частности, он способен подавлять транскрипцию многих других генов, например протоонкогенов BCL2, JUN и FOS, гена фибронектина и т. д. В основе такой транс-репрессии лежит несколько механизмов: связывание и секвестрация активированным р53 ряда базовых факторов транскрипции (p300/CBP, TBP, CBF); способность связывать и рекрутировать к определенным генам гистоновые деацетилазы (HDAC), ремоделирующие хроматин; и т. д. Кроме того, р53 связывается с белками, вовлеченными в репликацию или репарацию ДНК и, как следствие, модулирует эти процессы. Так, взаимодействуя с белком RP-A, он ингибирует его способность активировать ДНК-полимеразы a и d, результатом чего является подавление репликации ДНК. Связывая компоненты комплекса TFIIH (ERCC2, ERCC3 и др.), р53 активирует его функцию и стимулирует тем самым эксцизионную репарацию ДНК. Связывание р53 с белком Rad51 ведет к стимуляции рекомбинаций ДНК и повышению эффективности репарацию двунитевых разрывов ДНК. На участие р53 в репарации ДНК указывает также и его способность проявлять активность 3'-5'-экзонуклеазы и узнавать участки одноцепочечной ДНК и/или неспаренные основания.
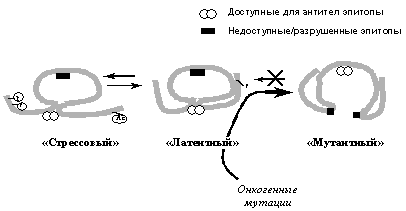
Рис. 3. Схематическое изображение различных конформационных состояний р53, распознаваемых специфическими антителами. Онкогенные мутации вызывают необратимый переход молекулы в денатурированное состояние, при котором открывается ранее недоступный эпитоп и, наоборот, исчезают некоторые ранее доступные эпитопы.
Характерные для опухолевых клеток миссенс-мутации приводят к резкому изменению конформации молекулы белка р53 (Рис. 3), что в значительной степени затрагивает все из вышеуказанных его активностей: происходит потеря или ослабление способности связывать и активировать гены с р53-респонсивными элементами, репрессировать другие специфические гены-мишени, ингибировать репликацию ДНК и стимулировать репарацию ДНК. Причем, так как р53 образует тетрамерные комплексы, мутации в одном аллеле гена р53 вызывают инактивацию и продукта второго, неповрежденного аллеля. Дело в том, что коэкспрессирующиеся нормальный и мутантный белок р53 образуют неактивные гетеромерные комплексы. Таким образом, мутантный белок ингибирует функции нормального белка р53 по доминантно-негативному механизму. По-видимому, именно эта особенность мутантных р53 в значительной мере ответственна за их онкогенный потенциал. В пользу этого свидетельствует тот факт, что введение в клетки короткого полипептида, соответствующего олигомеризационному домену р53, нарушает образование полноценных тетрамерных комплексов р53 и вызывает опухолевую трансформацию. Необходимо заметить, что помимо утраты нормальных функций р53, мутантные р53 с аминокислотными заменами в горячих точках (кодоны 175, 248 и др.) приобретают новые свойства, не свойственные белку р53 дикого типа (gain-of-function). Так, описано приобретение мутантными р53 способности активировать промоторы протоонкогенов MYC и ERB1, антиапоптотического гена BGL1 из семейства Bcl2, гена MDR1, детерминирующего множественную лекарственную устойчивость клеток и т. д. Предполагается, что это обусловлено способностью некоторых мутантных р53 связывать белки, в частности другие факторы транскрипции, с которыми нормальный р53 не взаимодействует, и модифицировать экспрессию генов, регулируемых этими транскрипционными факторами.
Физиологические функции р53 и их нарушения в неопластических клетках
Молекулы белка р53 могут находиться в различных конформационных состояниях, в которых они обладают разными биохимическими активностями и выполняют разные физиологические функции. В обычных условиях р53 находится в так называемой латентной форме, в которой он обладает слабой транскрипционной активностью. Такой р53, однако, связывает белки репарационной машины (см. выше), проявляет активность 3'-5'-экзонуклеазы и стимулирует рекомбинацию и репарацию ДНК. При различных стрессах и внутриклеточных повреждениях происходят пост-трансляционные модификации, в частности фосфорилирование и ацетилирование определенных аминокислот молекулы р53, определяющие ее переход в так называемую стрессовую конформацию. Такой р53 значительно более стабилен (т. е. резко увеличивается его количество в клетке) и эффективно транс-активирует и/или транс-репрессирует специфические гены-мишени, следствием чего является индукция в аномальных клетках либо остановки клеточного цикла, либо апоптоза. Кроме того, активация р53 ведет к изменению экспрессии генов некоторых секретируемых факторов, в результате чего может изменяться размножение и миграция не только поврежденной, но и окружающих клеток. При этом, находясь в стрессовой конформации, р53 в значительной степени утрачивает активности, стимулирующие рекомбинацию и/или репарацию ДНК.

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 |
