

Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Таблица 4.
Расчет и сравнение с экспериментом энергии растворения атомов гелия в вакансионном кластере (9 вакансий) с использованием потенциала IV
Число атомов гелия в кластере j |
|
|
|
|
0 | -5487.15976 | - | - | - |
1 | -5487.18569 | -0.02593 | -0.02593 | -0.028 ± 0.005 |
2 | -5487.19869 | -0.03893 | -0.01300 | -0.010 ± 0.003 |
3 | -5487.18421 | -0.02445 | +0.01448 | +0.050 ± 0.006 |
4 | -5487.13213 | +0.02763 | +0.05208 | +0.070 ± 0.007 |
Здесь E полная энергия системы, содержащей кластер и растворенный в нем гелий, 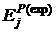 - экспериментальные значения энергии растворения (см. табл. 1). Погрешность определения полной энергии и остальных величин влияет только на последнюю значащую цифру.
- экспериментальные значения энергии растворения (см. табл. 1). Погрешность определения полной энергии и остальных величин влияет только на последнюю значащую цифру.
Моделирование растворения гелия в вакансионных кластерах разме-ром 8¸10 вакансий (потенциал IV) хорошо воспроизводит как энергии рас-творения первых двух атомов гелия, так и смену знака энергии растворения для третьего и четвертого. Для кластера из 8 вакансий величина энергии растворения четвертого атома гелия оказывается явно завышенной для обоих использованных потенциалов, тогда как растворение гелия в кластере из 9 вакансий дает хорошее совпадение величины энергии растворения третьего и четвертого атома с данными эксперимента (потенциал IV). По-видимому, конфигурация и размер реальных зернограничных вакансионных кластеров близки к модельному кластеру из 9 вакансий (рис. 4, c). Различие энергий растворения второго - четвертого атомов в одинаковых кластерах, но для разных потенциалов He-Pd (II и IV) обусловлено различной глубиной и протяженностью ямы этих потенциалов (см. рис. 3).
Смена знака с температурой для первой и второй ступеней (таблица 1) может быть обусловлена либо изменением конфигурации вакансионного кластера, в котором происходит растворение, с ростом температуры (например, отрыв вакансий от кластера), либо наличием других позиций растворения с повышенной энергией растворения в пределах кластера, заполняющихся только при более высокой температуре насыщения.
Полученные расчетные значения энергии растворения находятся в удовлетворительном согласии с экспериментальными величинами, что свидетельствует о применимости метода МД в решении подобных задач.
Показано, что минимальное значение экспериментальной энергии растворения обусловлено взаимодействием атома гелия с поверхностью свободного кластера. Зарегистрированное в МД эксперименте изменение энергии растворения гелия при увеличении числа атомов гелия в кластере находится в соответствии с предложенной моделью растворения.
На основе предложенной модели определена концентрация вакансионных кластеров, которая для данного образца составила ![]() см-3.
см-3.
Авторы благодарят проф. Мулюкова Р. Р. за предоставленные для исследования образцы палладия и Поташникова С. И. за расчет потенциала He-Pd методом МФП.
Список литературы
1. , , // ЖТФ. 1998. Т. 72. № 11. С. 96‑99.
2. Nazarov A. A. and Mulyukov R. R. Nanostructured Materials. In: Handbook of Nanoscience, Engineering, and Technology, Ed. Goddard W., Brenner D., Lyshevsk S., Iafrate G., CRC Press. 2002.
3. Mulyukov R. R., Starostenkov M. D. // Acta Met. Sinica. 2000. V. 13. № 1. P. 301‑309.
4. , , и др. // ФММ. 1986. T. 61. C. 1170.
5. М.: Металлургия. 19 С.
6. Würschum R., Kübler A. et. al. // Ann. de Chim. - Sci. des Mat. 1996. V. 21. P. 471‑482.
7. Купряжкин А.Я., Куркин А.Ю., Китаев Г.А. // ЖФХ. 1988. Т. 62. № 12. С. .
8. Wilson W. D., and Johnson R. A. “Rare gas in metals”, In Interatomic potentials and simulation of lattice defects. edited by P. C. Gehlen et. al., Plenum Press, New York – London, 1972, P. 375‑385.
9. Межфазовая граница газ - твердое тело. Под ред. Э. Флада. М.: Мир. 19 С.
10. Вараксин А. Н., Козяйчев В. С. // ФММ. 1991. № 2. С. 45-51.
11. Shu Zhen, Davies G. J. // Phys. stat. sol. (a). 1983. V. 78. № 72. P. 595‑605.
12. Гиршфельдер Дж., Молекулярная теория газов и жидкостей. М.: Издательство иностранной литературы. 19 С.
13. Michels, A., Wouters, H. // Physica. 1941. V. 8. P. 923
14. Delly B. // J. Chem. Phys. 1986. V. 92. P. 329.
15. Kim Y. S., Gordon R. G. // J. Chem. Phys. 1974. V. 61. № 1, P. 1-16.
16. Smith K. M., Rulis A. M., Scoles G. et. al. // J. Chem. Phys. 1977. V. 67. № 1, P. 152-163.
17. Zaremba E., Kohn W. // Physical rewiev B. 1977. V. 15. № 4, P. .
INTERACTION OF HELIUM WITH THE VACANCY CLUSTERS IN THE CRYSTALS Pd WITH SUBMICRON-GRAINED STRUCTURE
A. N. Zhiganov, A. Ya. Kupryazhkin
Urals state technical university - UPI, Ekaterinburg, Russia
Introduction
A study of interaction of helium with the structural materials of nuclear reactors is the topical task of reactor materials science, in particular, for the solution of the problem of the helium embrittlement of metals. There is independent interest in this case in the explanation of the nature of interaction of helium with the defects in the metal polycrystals and the mechanism of the formation of gas clusters. As the subject of the study in the present work the helium - palladium system with the submicron-grained structure was used. Palladium is a promising material of the membranes, used for isotope separation of hydrogen and their separation from helium. It can be model material for the structural materials, which have FCC structure.
1. Experimental results
Experimental research of the solubility of helium in palladium was performed on the installation, described in [1], by the method of the thermal desorption of helium from the previously saturated in the helium atmosphere sample with the required temperature of saturation T and saturation pressure P. After that the sample was transferred from the saturation chamber to the measurement chamber. The solubility of helium in palladium was calculated from the data on complete desorption of the sample. Error of solubility determination was not more than 10%.
The samples of polycrystalline Pd with purity 99.99% were kindly submitted for researches by prof. R. R. Mulyukov. The submicron-grained structure of sample was obtained with the aid of the large plastic deformation by the method of twisting under the quasihydrostatic pressure on the installation of the type of the Bridgeman anvil [2, 3, 4]. Plastic deformation is characterized by the true logarithmic degree e = ln(lk/l0), where l0 и lk are initial and finite dimensions [5]. In our case e=7. It follows from the electron-microscopic examinations, carried out with the aid of the transmission electron microscope JEM 2000EX, that the samples acquire the strongly dispersed structure with average size of grains of 150 nm [6] as a result of intensive plastic deformation. The dislocation density in the sample is ~3·1010 cm-2.
As result of the study the dependences of the solubility of helium on the saturation pressure Ceff=f(P) on the solubility curve four “plateau” (stages) are registered, which characterize saturation of defects (Fig. 1). The values of Ceff for the appropriate “plateau” denote ![]() , (N = 1÷4). For the convenience only a part of the registered isotherms is shown on the figure. It is discovered, that for the solubility isotherms with a different temperature of saturation for “plateau” with the identical numbers have value
, (N = 1÷4). For the convenience only a part of the registered isotherms is shown on the figure. It is discovered, that for the solubility isotherms with a different temperature of saturation for “plateau” with the identical numbers have value ![]() identical within the limits of experimental error. This implies that saturation of the positions of the dissolutions occurs, whose concentration does not depend on temperature.
identical within the limits of experimental error. This implies that saturation of the positions of the dissolutions occurs, whose concentration does not depend on temperature.
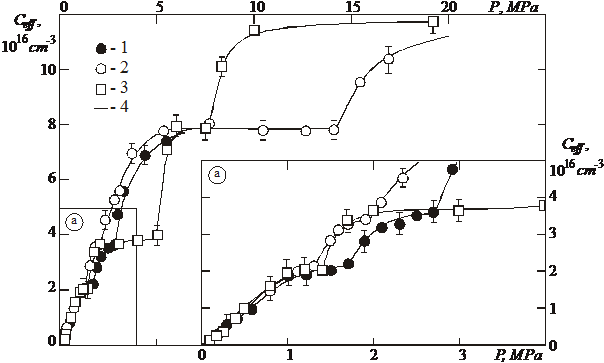
Fig. 1. Solubility isotherms of helium in polycrystalline palladium with submicron-grained structure. 1 – Т=387 K; 2 ‑ Т=273 K; 3 - Т=483 K; 4 ‑ solid lines - approximation.
It follows from shape of dependence 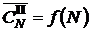 , averaged on the saturation temperature (Fig. 2), that the dependence can be described by the linear function, passing through the origin of coordinates (
, averaged on the saturation temperature (Fig. 2), that the dependence can be described by the linear function, passing through the origin of coordinates (![]() ). On the basis of this it is possible to assume that saturation of one and the same positions of dissolution occurs, and, at the low saturation pressure these positions are filled with one atom of helium (first “plateau”). Then, with an increase in the saturation pressure, a second helium atom is added in the same position to the already dissolved atom (the second “plateau”). The third and the fourth “plateau” are formed in the analogous way.
). On the basis of this it is possible to assume that saturation of one and the same positions of dissolution occurs, and, at the low saturation pressure these positions are filled with one atom of helium (first “plateau”). Then, with an increase in the saturation pressure, a second helium atom is added in the same position to the already dissolved atom (the second “plateau”). The third and the fourth “plateau” are formed in the analogous way.
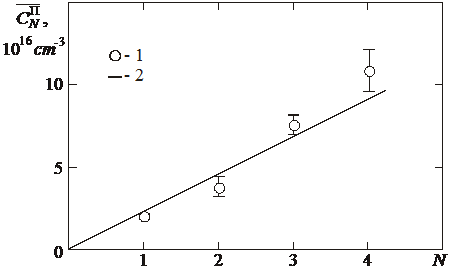
Fig. 2. Dependence ![]() on the “plateau” number N. 1 - experimental points; 2 - approximation.
on the “plateau” number N. 1 - experimental points; 2 - approximation.
It is evident from Fig. 2 that all the stages have clearly shown origin (linear dependence of solubility on the saturation pressure) and extensive “plateau”. The region of linear dependence on the saturation pressure of each stage begins at the well determined saturation pressure, when an increase in the solubility in the “plateau” of the previous stage is already insignificant. This means that filling of dissolution positions by (j+1)-th helium atom begins at the concrete saturation pressure only after complete saturation of the dissolution positions by the j-th helium atom. Thus, it is possible to write (for example, see [7]) expression for the solubility dependence on the saturation pressure for the j‑th (j=1¸4) stage for all isotherms in the form
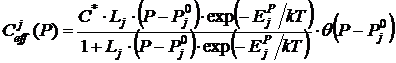 . (1)
. (1)
Here j is the number of stage; ![]() cm-3 is the concentration of the dissolution positions;
cm-3 is the concentration of the dissolution positions; ![]() is the dissolution energy of the j-th atom of helium;
is the dissolution energy of the j-th atom of helium; ![]() is pressure, at which the stage with the number j begins; q (x) = 1 (x³0), q (x) = 0 (x<0);
is pressure, at which the stage with the number j begins; q (x) = 1 (x³0), q (x) = 0 (x<0); ![]() are constants with relatively weak dependence on T and P.
are constants with relatively weak dependence on T and P.
Because of the assumption of the independent filling of the saturation positions with each new helium atom (at any stage of isotherm), the dependence of solubility on the pressure at the low saturation pressure (relative to the beginning of stage) must obey the law of Henry, i. e., the proportionality band in which the solubility is proportional to saturation pressure and exponentially depends on reverse temperature (1) must be in the beginning of each stage. Treatment of the proportionality band of each stage makes it possible to obtain the energy of dissolution ![]() , which corresponds to the dissolution of the sequential (j-th) atom of helium in the dissolution positions, which contain (j‑1) helium atoms.
, which corresponds to the dissolution of the sequential (j-th) atom of helium in the dissolution positions, which contain (j‑1) helium atoms.
It is discovered from the treatment of the dependences of solubility on saturation temperature, that these dependences for first two stages consist of two regions: low-temperature region (273¸400 K) and high-temperature region (400¸508 K), for each of which it is possible to obtain a dissolution energy. The values of all the obtained dissolution energies are shown in Table 1.
It is shown from the study of grain size with the annealing of identical samples in the work [6] that in the range of temperatures (293¸508) K their size increases by more than 2, and therefore the concentration of the helium dissolution positions within the grains boundaries, created in the process of the sample deformation, significantly decreases. However, the results obtained in the experiment do not depend on this.
Table 1.
Parameters of the temperature dependence ![]() of helium in Pd in the regions with the linear dependence Cjeff = f(P-P0j)
of helium in Pd in the regions with the linear dependence Cjeff = f(P-P0j)
Stage | DT, K | Dependence parameters | |
|
| ||
1. | 273¸400 |
| -0.028 ± 0.005 |
400¸508 |
| +0.086 ± 0.009 | |
2. | 293¸430 |
| -0.010 ± 0.003 |
430¸508 |
| +0.077 ± 0.009 | |
3. | 293¸508 |
| +0.050 ± 0.006 |
4. | 293¸508 |
| +0.070 ± 0.007 |
According to results calculated by the lattice static method [8], the helium dissolution energy in the Pd vacancy have value ![]() =0.52 eV, and in the interstitial position
=0.52 eV, and in the interstitial position ![]() =3.68 eV. These values substantially exceed the obtained experimental values of helium dissolution energy (Table 1). Consequently, vacancy and interstitial position should be excluded from the assumed positions of dissolution, because their contribution to the solubility must be negligible. An analogous conclusion can be made about dislocations and small-tilt grain boundaries, which consist of the dislocation walls; helium dissolution energy in these positions is comparable with the dissolution energy in the vacancies.
=3.68 eV. These values substantially exceed the obtained experimental values of helium dissolution energy (Table 1). Consequently, vacancy and interstitial position should be excluded from the assumed positions of dissolution, because their contribution to the solubility must be negligible. An analogous conclusion can be made about dislocations and small-tilt grain boundaries, which consist of the dislocation walls; helium dissolution energy in these positions is comparable with the dissolution energy in the vacancies.
On the other hand, the experimental data on helium interaction with the solid surface [9] gives the negative values of adsorption energy of helium on the surface in the range (-0.026¸-0.029) eV. The values are compared with the energy of the corresponding low-temperature region of solubility curve (Table 1, stage 1) for the proportionality band of first stage (“plateau”) ![]() =(‑0.028±0.005) eV, that indicate the physical adsorption of the helium atoms at the free internal surface of the sufficiently large (of ³8 vacancies) vacancy clusters localized, apparently, near the large-tilt grain boundaries which not being annealed at study temperature.
=(‑0.028±0.005) eV, that indicate the physical adsorption of the helium atoms at the free internal surface of the sufficiently large (of ³8 vacancies) vacancy clusters localized, apparently, near the large-tilt grain boundaries which not being annealed at study temperature.
For testing the proposed dissolution mechanism the simulation of the helium atom dissolution in the vacancy cluster which consists of 8¸10 vacancies by the molecular dynamics method (MD) was conducted.
2. Simulation of the helium dissolution in palladium
Simulation of the helium dissolution in palladium was performed with the aid of the molecular dynamics method. During the simulation, the particles were represented in the form of material points of the masses equal to the atom masses. Interaction of the particles was described by the pair central potentials, given either parametrically in the known functional form or by table with further spline-interpolation during the simulation. For describing the motion of classical particles the system of the differential equations of Newton's dynamics was used in the finite-difference form, which were solved by the Euler's method with half-step (2). The initial conditions of task are determined by the initial configuration of system (coordinates, impulses).
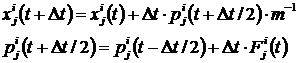 . (2)
. (2)
Here x, p are coordinates and impulses of the particle in system at the appropriate moment of time, Dt is step of integration for the time, index i is the number of the particle, iÎ[1..N], where N is number of particles of the system, the index jÎ[1..3] is number of component. Force F is the composite force, which acts on the particle with number i from remaining particles of the system and is calculated according to formula (3).
![]() . (3)
. (3)
Where j is pair potential of interaction between particles with numbers i and k.
The periodic boundary conditions were used, the step of integration for the time has value Dt=3×10-15 s. For the simulation of the helium dissolution of in the palladium crystal the system of 1372 palladium atoms with cubic crystalline shape and size of the cube edge was equal 7 periods of the unit cell (a0=0.388 nm).
The choice of the pair central potentials of interaction for all pairs of the particles types of the system was performed on the basis of the analysis of the accessible potentials found in the literature. So, potential Pd-Pd is taken from the works [10, 11], potential He-He - from the works [12, 13]. The choice of potential He-Pd was the basic problem for the simulation. Only one accessible potential obtained by the density functional method, and only in the region of repulsion (I) had been found in the work [8]. One additional potential (II), which extends to the attraction region and has a depth of the well e=1.993 meV, was calculated with the aid of the original program package developed by us for pairwise interactions calculation on the basis of the density functional method. Potentials I and II give a good agreement in the region of repulsion. Potential obtained by the quantum-chemical Dmol method [14] strongly differs from the potentials I and II in the repulsion region and has anomalously deep well e=74.76 meV. Potential III, which is a modification of potential I obtained by adding of the dispersion interaction calculated by the Gordon - Kim technique [15] with the well e=2.50 meV also was used. Potential IV is other modification of the potential I with dispersion interaction added to it; here the addition was done in the HFD1 form of the Buckingham potential [16], which considers damping dispersion interaction at short distances. The depth of the potential IV well is e=1.04 meV. All the potentials (except Dmol) are given at Fig. 3.
The criterion of the applicability of the potentials for the simulation of the interaction between helium and palladium is the proximity of dissolution energies of the helium atom in vacancy and interstitial positions of Pd lattice to the values obtained by authors [8]. Additional criterion is value of bond energy of the helium atom with the surface of metals (plane {111}) comparable with the calculations MFP in the “jelly” model. Because the data for the system He‑Pd are not available, the data for system He-Au were used. The maximum value of the bond energy of the helium atom with the surface of gold has value EA= ‑ 8 meV [17].
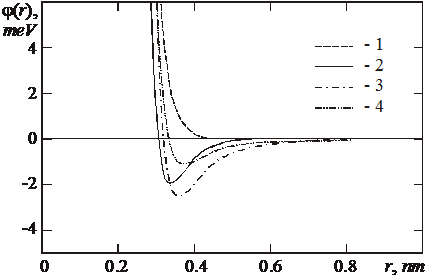
Fig. 3. Pair central potentials of interaction He-Pd. 1 - potential I; 2 - potential II; 3 - potential III;
4 - potential IV (explanation in the text).
The parameters of the dispersion interaction in the potential IV had been chosen so as to reproduce adsorption energy on the surface of gold. The data of the potentials comparison are shown in table 2. It is evident that all the dissolution energies in the vacancies and interstitial positions are smaller than obtained in the work [8]. This can be explained by the fact that the potential Pd‑He used in [8] has only a repulsion term, whereas all remaining potentials (II - IV) have attracting component (see fig. 3). A difference between the results of [8] and our calculation with the potential He-Pd from [8] and from [11] for the helium dissolution energy in vacancies and interstitial positions of palladium is explained by the differences in the used Pd-Pd potential, that gives different contribution of relaxation energy of the of palladium lattice near the dissolved helium atom to the dissolution energy. In further calculations of the energy of the helium dissolution in the vacancy clusters of different configuration potentials II and IV were used, because the potential I does not have attraction, but the potential III gives the too high EA energy.
Table 2.
Comparison of dissolution ![]() ,
, ![]() and adsorption EA energies for the used pair potentials of the He-Pd interaction
and adsorption EA energies for the used pair potentials of the He-Pd interaction
potential | potential |
|
| EA, meV |
Data [8] | 0.52 | 3.68 | - | |
I [8] | [11] | 0.36 | 3.30 | - |
II | 0.16 | 2.98 | -7.5 | |
III | 0.30 | 4.12 | -18.9 | |
IV | 0.25 | 3.17 | -7.8 |
Before the simulation, the initial configuration of the system containing the vacancy cluster of the corresponding structure and 1 to 4 dissolved helium atoms located in it, was created. Impulses of the system particles were chosen random in the direction so that the temperature of system would be equal to the required temperature at the beginning of simulation (100 K). During further simulation, the necessary number of steps with the continuous cooling of system was performed. The relaxation of the lattice near the vacancy cluster and the arrangement of the helium atoms in the cluster with the minimum of dissolution energy occur during the cooling process. The duration of simulation was chosen such that the temperature of system would decrease to the value, which gives the contribution to the total energy of system (expressed in eV) at the degree of 5 decimal places. For the system of 1372 atoms the duration of the simulation was 5000 time steps; the temperature of the system at the end of the simulation was equal 3×10‑5 K. The total energy of system was calculated by the averaging over the last 500 time steps of the total energy values at time step.

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 |
