
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
вещества вторичного обмена
При исследовании веществ вторичного обмена первоначально двумерной БХ мы проанализировали качественный состав фенольных соединений надземных органов вереска обыкновенного. При этом нами обнаружено 18 веществ флавоноидной природы, среди гидроксикоричных кислот – не менее 8 веществ, из которых по окраске до и после проявления, по значению Rf при сравнении с достоверными образцами предварительно идентифицировали кверцетин, гиперозид и хлорогеновую кислоту. Причем на основании площади пятен и интенсивности их окрасок после проявления диагностическими реактивами отмечено, что они являются доминирующими фенольными соединениями побегов вереска.
Уточнение данных двумерной хроматографии гидроксикоричных кислот провели на отечественном жидкостном микроколоночном хроматографе «Милихром-4» НПО «Научприбор» (г. Орел), в ходе которого полностью подтвердили, что преобладающей гидроксикоричной кислотой побегов вереска являлась хлорогеновая (табл. 3, рис. 1). Кроме нее, в весьма значительных концентрациях определены хинная и кофейная кислоты, в минимальной концентрации обнаружена феруловая кислота.
| Рис. 1. Хроматограмма ВЭЖХ-анализа побегов вереска Условные обозначения гидроксикоричных кислот: 1 – хлорогеновая, 2 – кофейная, 3 – феруловая, 4 – хинная. |
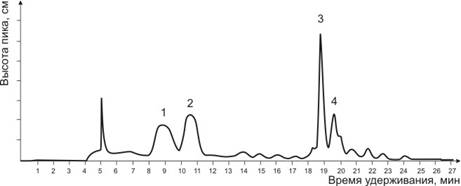
Таблица 3 – Содержание гидроксикоричных кислот в побегах вереска обыкновенного
Кислота | Содержание в образце, мг/мл извлечения | Кислота | Содержание в образце, мг/мл извлечения |
Хинная | 0,22 | Феруловая | 0,02 |
Кофейная | 0,12 | Хлорогеновая | 0,45 |
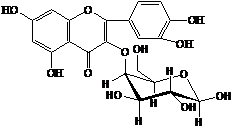
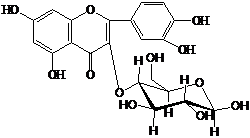
Для подтверждения данных двумерной хроматографии провели выделение флавоноидов в индивидуальном состоянии колоночной хроматографией на полиамиде с дальнейшим их физико-химическим изучением. Так, при исследовании выделенных веществ в УФ-области спектра в присутствии ионизирующих и комплексообразующих реагентов и анализе их ПМР-спектров вещество I охарактеризовали как 3,5,7,4’-тетраоксифлавон (кемпферол), вещество II – как 3,5,7,3’,4’-пентаоксифлавон (кверцетин), вещество III – как 3-О-b-D-глюкозид кемпферола (астрагалин), вещество IV – как 3-О-b-D-галактозид кверцетина (гиперозид), вещество V – как 3,5,7,8,4’-пентаоксифлавон (гербацетин), вещество VI – как 8-О-b-D-глюкозид гербацетина и вещество VII – как 3-О-b-D-глюкозид кверцетина (изокверцитрин). Их структурные формулы отражены в таблице 4.
Таблица 3 – Структурные формулы отдельных флавоноидов побегов вереска
Вещество I – 3,5,7,4’-тетраоксифлавон (кемпферол) |
Вещество II – 3,5,7,3’,4’-пентаоксифлавон (кверцетин) |
Вещество III – 3-О-b-D-глюкозид кемпферола (астрагалин) |
Вещество IV – 3-О-b-D-галактозид кверцетина (гиперозид) |
Вещество V – 3,5,7,8,4’-пентаоксифлавон (гербацетин) | |
Вещество VI – 8-О-b-D-глюкозид гербацетина |
Вещество VII – 3-О-b-D-глюкозид кверцетина (изокверцитрин) |
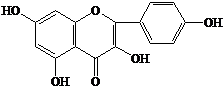
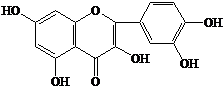

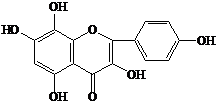

Для выявления и подтверждения преобладающих компонентов суммы гидроксикоричных кислот и суммы флавоноидов провели сравнение спектров поглощения для гидроксикоричных кислот извлечения побегов вереска и стандартов кислот (кофейной, феруловой, хлорогеновой), для флавоноидов – комплексов извлечения вереска, кверцетина-стандарта и гиперозида-стандарта с алюминия хлоридом. Совпадение максимумов спектров поглощения извлечения вереска и стандартов хлорогеновой кислоты и кверцетина (рис. 2 и 3) позволили использовать их в качестве стандартных веществ.

Рис. 2. УФ-спектры поглощения спиртового извлечения побегов вереска
и стандартов гидроксикоричных кислот
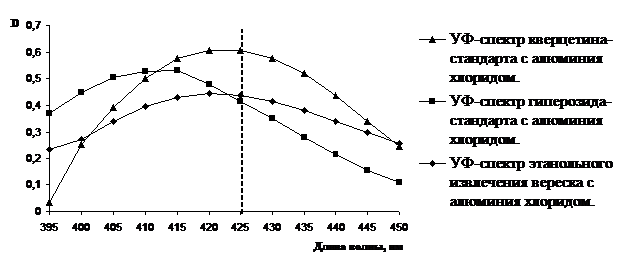
Рис. 3. УФ-спектры поглощения комплексов кверцетина-стандарта, гиперозида-стандарта
и этанольного извлечения побегов вереска обыкновенного с алюминия хлоридом
количественное определение фенольных соединениЙ
Фенологликозиды, гидроксикоричные кислоты, флавоноиды и полифенольные окисляемые соединения – одни из важнейших групп действующих веществ побегов вереска, для стандартизации которых мы предлагаем проводить их количественное определение. При этом нами разработаны методики количественного определения сумм флавоноидов, гидроксикоричных кислот, полифенольных окисляемых соединений, включающие такие параметры как экстрагент и условия экстракции, а также арбутина с использованием хроматографической очистки извлечения и удельного показателя поглощения арбутина-стандарта.
Гидроксикоричные кислоты. Сумму гидроксикоричных кислот определяли методом прямой спектрофотометрии. Оптимальные условия экстракции следующие: экстрагент – 90% спирт этиловый, время и кратность экстракции – 4 раза в течение 30 минут. В качестве стандарта использовали хлорогеновую кислоту производства фирмы «Fluka» (Германия), удельный показатель поглощения которой равнялся 504,425.
Метрологическая характеристика количественного определения суммы гидроксикоричных кислот методом прямой спектрофотометрии приведена в таблице 5. Ошибка определения находилась в пределах ±1,58%.
Таблица 5 – Метрологическая характеристика количественного определения гидроксикоричных кислот
n | f | X | S2 | S | Sx | P,% | t (p, f) | ΔX | ε,% |
10 | 9 | 2,148 | 0,0022 | 0,046 | 0,015 | 95 | 2,26 | 0,034 | 1,58 |
Результаты количественного определения гидроксикоричных кислот в побегах вереска обыкновенного из различных мест произрастания отражены в таблице 11. В ходе исследования отметили, что содержание гидроксикоричных кислот варьировало в пределах от 2,343±0,0384% до 9,139±0,1414%. Их минимальные концентрации нами выявлены в образцах, заготовленных в окр. г. Брянска, г. Витебска, отдельных местах произрастания в Ярославской области; а наиболее высокие – в образцах из окр. пос. Смирдомский Чагодощенского района Вологодской области, г. Мурманска, Республики Коми, г. Петрозаводска и других.
Флавоноиды. Для количественного определения суммы флавоноидов в побегах вереска использовали спектрофотометрическую методику. В ее основу положили реакцию комплексообразования с алюминия хлоридом в сочетании со спектрофотометрическим определением оптической плотности комплексов в видимой области, что позволило проводить непосредственное спектрофотометрирование в извлечениях из сырья без дополнительных трудоемких стадий очистки. Оптимальные условия экстракции следующие: экстрагент – 70% спирт этиловый, время и кратность экстракции – 3 раза по 15 минут. Удельный показатель поглощения комплекса кверцетин - алюминия хлорид равнялся 596,1.
Метрологическая характеристика количественного определения суммы флавоноидов по упомянутой методике приведена в таблице 6. Ошибка определения суммы флавоноидов в среднем составила 1,61%.
Таблица 6 – Метрологическая характеристика количественного определения флавоноидов
n | f | X | S2 | S | Sx | P,% | t (p, f) | ΔX | ε,% |
10 | 9 | 0,807 | 0,00033 | 0,0182 | 0,0058 | 95 | 2,26 | 0,013 | 1,61 |
Результаты количественного определения флавоноидов в побегах вереска, собранных в различных местах произрастания, обобщены в таблице 11. При этом выявили, что содержание суммы флавоноидов варьировало от 0,413% до 1,386%. Ее минимум приходился на образцы, заготовленные в окр. г. Рыбинска, в окр. местечка Симак, в окр. пос. Нижний, д. Кобостово, д. Малый Солонец Ярославской области, окр. г. Брянска и др., а максимальное содержание – на образцы, собранные в окр. г. Петрозаводска Республики Карелия, окр. г. Вохи Кирилловского района Вологодской области, окр. г. Мурманска, окр. санатория «Малые соли» Некрасовского района Ярославской области, окр. с. Унжа Койгородского района Республики Коми и др.
полифенольные окисляемые соединения (ПОС). Их количественное определение проводили по методике ГФ XI перманганатометрическим титрованием в присутствии индигосульфокислоты (метод Левенталя в модификации ).
Метрологическая характеристика количественного определения ПОС приведена в таблице 7. Ошибка их определения составила в среднем 3,23%. Результаты определений отражены таблице 11.
Таблица 7 – Метрологическая характеристика количественного определения полифенольных окисляемых соединений
n | f | X | S2 | S | Sx | P,% | t (p, f) | ΔX | ε,% |
10 | 9 | 6,590 | 0,089 | 0,298 | 0,094 | 95 | 2,26 | 0,213 | 3,23 |
В ходе исследования обнаружили, что содержание ПОС находилось в пределах от 2,410% до 9,406%, причем их максимум отметили в образце из окр. г. Брянск Брянской области, а минимум – в образце из окр. с. Ужга Койгородского района Республики Коми.
арбутин. В отличие от методики ГФ XI издания содержание арбутина определяли прямой хромато-спектрофотометрией с использованием стандарта арбутина фирмы «Sigma» (США). Его максимум поглощения нами отмечен при длине волны 285±2 нм. Он совпадал с максимумом спиртового извлечения побегов вереска (рис. 4).
Вначале мы определили удельный показатель арбутина-стандарта (табл. 8), который составил 72,23 и в дальнейшем использовался при количественном определении.
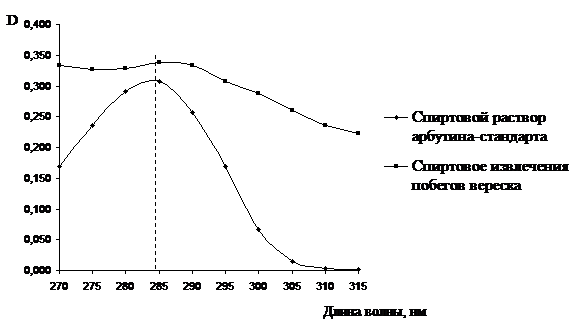
Рис. 4. Спектры поглощения спиртового извлечения побегов вереска обыкновенного и
арбутина-стандарта
Таблица 8 – Определение удельного показателя поглощения арбутина-стандарта
Концентрация арбутина, % | D |
| Метрологическая характеристика |
0,001 | 0,082 | 82,00 | `x = 72,23 s = ±4,26 s`x = ±3,32 I0,95 = ±2,23 M = 72,23±2,23 d = 1,285 |
0,002 | 0,149 | 74,50 | |
0,003 | 0,229 | 76,33 | |
0,004 | 0,296 | 73,25 | |
0,005 | 0,358 | 73,30 | |
0,006 | 0,426 | 70,59 | |
0,007 | 0,484 | 69,14 | |
0,008 | 0,558 | 69,00 | |
0,009 | 0,633 | 70,33 | |
0,010 | 0,684 | 68,40 | |
0,011 | 0,744 | 67,64 |
Оптимальные условия экстракции для арбутина следующие: экстрагент – 70% спирт этиловый, время и кратность экстракции – 3 раза по 30 минут.
Очистку от сопутствующих веществ (другие фенольные соединения) мы проводили с использованием колонки с сорбентом – алюминия оксидом. Учитывая, что при проведении хроматографической очистки часть арбутина задерживалось алюминия оксидом, мы определили коэффициент неполного элюирования (табл. 9), который составил 1,14095.
Таблица 9 – Определение коэффициента неполного элюирования
Опыт | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
Исходная концентрация раствора арбутина, % | 0,05 | 0,05 | 0,05 | 0,05 | 0,05 | 0,05 | 0,05 | 0,05 |
Полученная концентрация, % | 0,0424 | 0,0443 | 0,044 | 0,0431 | 0,0429 | 0,046 | 0,433 | 0,0448 |
Метрологическая характеристика | `x = 1,14095 σ = ±0,03 | М = 1,14095±0,03 δ = 0,011 |
Метрологическая характеристика количественного определения арбутина методом прямой спектрофотометрии приведена в таблице 10. Ошибка определения равнялась 1,010%. Результаты анализа отражены в таблице 11.

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 |
