

Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Присутність циклічних поліестеретерів з невисоким ступенем поліконденсації у продуктах отриманих при синтезах на основі Glu(Boc) та Glu(Ac) було підтверджено з використанням мас-спекроскопії. Так, спостерігаються сигнали циклічних кополіестерів зі ступенем поліконденсації 1-3. Треба зауважити, що їх не було виявлено у випадку використання Glu(St) та Glu(L).
Разом з тим, визначені кількості циклічних кополіестерів, у реакційній суміші, є недостатнніми, для пояснення величини неспівпадіння se та sc.
Систематичні дослідження кополіестерів методом мас-спектроскопії дозволили виявити макромолекули з різним ступенем кополіконденсації, з кінцевими групами відмінними від карбоксильних або гідроксильних незалежно від природи Д-α-АК, і природи реагенту який при синтезі був використаний в надлишку.
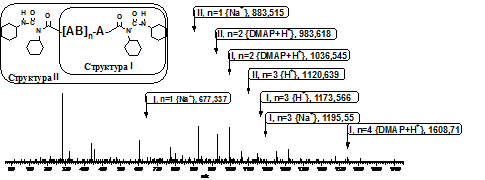 Рис. 3. Мас-спектр продуктів отриманих при синтезі ко-Glu(Ac)-ко-DЕG (температура 303К, низькі концентрації). Віднесення сигналів фрагментів з пасивними кінцевими групами.
Рис. 3. Мас-спектр продуктів отриманих при синтезі ко-Glu(Ac)-ко-DЕG (температура 303К, низькі концентрації). Віднесення сигналів фрагментів з пасивними кінцевими групами.
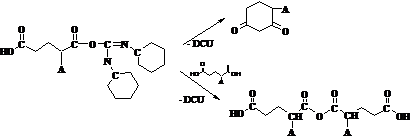
Можна зробити висновок, що при перебігу реакції частково відбувається прегрупування активованої DCC карбоксильної групи в пасивну групу N-ізоацилсечовини (схема 3) і ланцюг поліконденсації обривається.
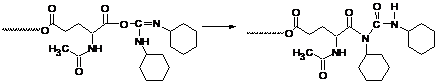 |
Схема 3. Реакція перегрупування активованої DCC карбоксильної групи в нереакційноздатну кінцеву групу N-ізоацилсечовини.
На рис. 3 наведено мас-спектр, який ілюструє присутність в макромолекулах пасивних кінцевих груп N-ацилсечовини, які є наслідком перегрупування.
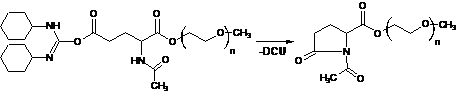 |
Схема 4. Деактивування активованої форми карбоксильної групи через внутрішньомолекулярну циклізацію з утворенням піролідонового циклу.
Окрім того, аналіз масспектрів продуктів дозволив виявити також перебіг побічного перетворення активованої форми карбоксильної групи через внутрішньомолекулярну циклізацію, згідно схеми 4, з утворенням кінцевих груп неактивних в поліконденсації, які можна розглядати, як похідні піролідону. Перебіг реакції утворення циклічних поліетерестерів та реакція внутрішньо молекулярної циклізації (схема 4) супроводжуються виділенням DCU. Пониження ступеня поліконденсації при тому супроводжується збільшенням різниці між sе і sc. Невідповідність між sc і sm, на величину більшу ніж це зумовлює конверсія процесу
визначається непродуктивним перегрупуванням активованої форми карбоксильної
 |
групи (схема 3) з утворенням кінцевих груп N-ацилсечовини, оскільки цей процес протікає без виділення DCU, за кількістю якої проводилась оцінка конверсії. Враховуючи те, що частка утворених циклічних поліестеретерів є незначною, то аналіз співвідношення між величиинами se, sc, sm дозволяє оцінити яка з побічних реакцій має максимальний вплив на пониження ступеня кополіконденсації в залежності
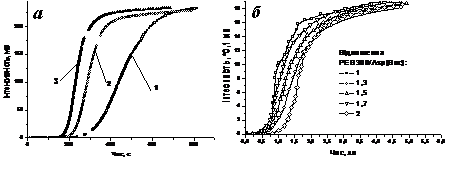
Рис. 4. Кінетичні криві виділення DCU в ході кополіконденсації Д-α-АК з PEG300 при 288К при різних а) початковій концентрації Glu(St) (1-0,0165 моль/дм3, 2-0,0219 моль/дм3, 3-0,0253 моль/дм3) та б) початковій концентрації концентрації PEG300 при поліконденсації з Asp(Boc).
від природи N-замісника в Д-α-АК та умов проведення процесу. В більшості випадків синтезів, в тому числі для оптимальних умов і структур (винятком є синтези 17,18 табл. 1), відбуваються обидві побічні реакції. У випадку синтезу низькомолекулярних естерів, перебіг побічних реакцій пригнічують збільшенням концентрації (надлишку) спирту. Проте, такий шлях є неможливим для випадку поліконденсації. Неефективним виявилось використання і супресора перегупування - N-гідроксибензотріазолу. Проведені дослідження показали, що в цьому випадку для забезпечення швидкого перебігу реакції температура повинна перевищувати 373К.
Нами встановлено, що природа N-замісника в Д-a-Ак має вирішальний вплив на ефективність перебігу поліконденсації за реакцією Стегліха. Досягнення максимального ступеня поліконденсації для похідних Д-α-АК з об’ємними N-замісниками дозволяє стверджувати, що процес утворення циклічних естерів в цьому випадку практично не відбувається. Для таких реагентів з усіх вище описаних побічних перетворень, в помітній мірі протікає тільки деактивація через утворення піролідонового циклу за схемою 4.
Таким чином, тільки у випадку поліконденсації за реакцією Стегліха N-похідних Д-a-АК з об’ємними замісниками в N-положенні ступінь поліконденсації можна було оцінювати за рівнянням Карозерса. У інших випадках, в тому числі для лабільних замісників, реакція естерифікації протікає менш ефективно, і реалізується лише на 24÷65% від визначеного співвідношенням реагентів та конверсією.
За виділенням DCU, крім визначення ступеня перетворення, можна оцінити динаміку процесу поліконденсації. На рис.4 наведені кінетичні криві виділення DCU в реакційній суміші. Методика нефелометричної оцінки дозволяє фіксувати Схема 5. Утворення естерної групи в результаті нуклеофільної атаки гідроксильною групою активованої DCC форми карбоксильної групи.
виділення DCU до конверсії 60÷75%.
 |
S-подібний характер кінетичних кривих вказує на те, що DCU виділяється в результаті перебігу ряду послідовних реакцій. Аналіз кривих (рис. 4.а) показує, що концентрація Д-α-АК, яка визначає кількість Схема 6. Реакції утворення ангідридих груп за участю активованої форми карбоксильної групи в реакції Стегліха.
карбоксильних груп в реакційній суміші суттєво впливає на швидкість виділення DCU. З кривих рис.4.б видно, що концентрація спиртової компоненти до значної (не менше 68÷77%) конверсії практично не впливає на швидкість реакції. Більш детальний аналіз кінетичних кривих, для ряду Д-α-АК, при різних початкових концентраціях діолів показав, що їх концентрація впливає на швидкість виділення DCU лише на глибоких стадіях перетворення.
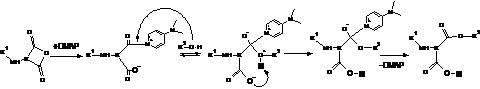
Схема 7. Перетворення ангідридної групи в естерну в реакції Стегліха в присутності DMAP.
Аналогічні дослідження з використанням масс-спектроскопії показали, що концентрація активатора (DCC) та нуклеофільної добавки (DMAP) також не визначають швидкості виділення DCU практично на всіх стадіях реакції кополіконденсації. Таким чином, можна зробити висновок, що DCC та DMAP не приймають участі в стадіях, які лімітують процес, а вплив спиртової компоненти проявляється лише на глибоких стадіях перетворення. Крім того, одержані кінетичні криві показують, що основна кількість DCU (до 60÷70%) виділяється відносно швидко (за всіх умов не більше 5÷7 хв). Після цього виділення DCU значно сповільнюється і для досягнення високої ступені поліконденсації, рекцію необхідно проводити ще на протязі 4÷5 годин.
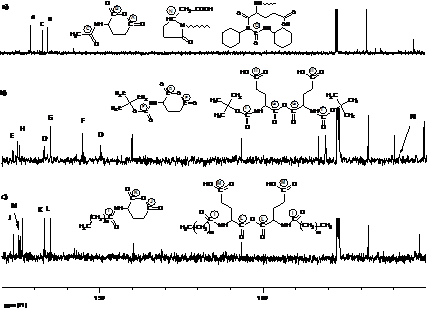
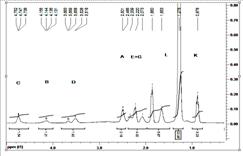
Рис. 5. ЯМР-С13 спектри модельної реакційної суміші на основі а) Glu(Ac), б) Glu(Boc), с) Glu(L).
Пояснення фактам, які спостерігаються в ході описаних кінетичних досліджень, можна зробити виходячи з особливостей механізму реакції Стегліха. Естерна група може утворюватись в результаті взаємодії активованої форми карбоксильної групи по реакції за схемою 5. За цією схемою реакція проходить у випадку синтезу низькомолекулярних естерів при значному надлишку в реакційній суміші спирту. Разом з тим, відомо про можливість перебігу реакції Стегліха через проміжну стадію одержання ангідридів. Для системи, що досліджується, такий процес може бути відображений реакціями, що наведені на схемі 6. Використання двоосновної кислоти, припускає можливість утворення як внутрішньомолекулярного, так і міжмолекулярного ангідриду. Імовірність утворення ангідриду, особливо велика на початкових стадіях процесу (до виділення 50% DCU). Подальше перетворення ангідридних груп в естерні в присутності DMAP може бути відображено реакціями схеми 7.
Перебіг реакції поліконденсації за реакцією Стегліха через проміжну стадію утворення ангідридів дозволяє пояснити кінетичні закономірності, які спостерігаються експериментально за виділенням DCU. Дійсно якщо стадією, яка лімітує процес, є утворення ангідридної групи, тоді поліетердіоли, DMAP, DCC не приймають в ній участі і їх концентрація не впливає на швидкість. В такому випадку швидкість реакції, визначається лише концентрацією карбоксильних груп, оскільки саме вони визначають концентрацію їх активованої форми. У ході вичерпування карбоксильних груп та збільшення молекулярної маси макромолекул кополіестерів, на глибоких стадіях перетворення, з реакцією утворення ангідридних груп починає конкурувати реакція утворення естерних груп за схемою 5. В результаті цього, на глибоких стадіях стає помітним вплив концентрації спирту. Але разом з тим, конкурентними до реакції утворення ангідридних груп стають і побічні реакції які приводять до дезактивації активованої форми карбоксильної групи згідно схем 3 та 4. В результаті протікання цих реакцій відбувається обрив як матеріального так і кінетичного ланцюга реакції поліконденсації.
Перебіг реакції поліконденсації через проміжне утворення ангідридних груп, було підтверджено С13 ЯМР дослідженнями у модельних реакційних сумішах, отриманих в умовах кополіконденсації, але в які не вводилася спиртова компонента. Завдяки цьому забезпечувалась можливість накопичення ангідридних груп в кількостях достатніх для реєстрації сигалів від атомів карбону з їх складу. Характерні С13 ЯМР спектри з віднесенням основних сигналів для різних N-похідних Д-a-АК наведені на рис.5. В спектрах добре ідентифікуються характерні сигнали, як від внутрішньомолекулярних ангідридних груп (А, В) так і характерні сигнали міжмолекулярних ангідридних груп (G). Отримані результати С13 ЯМР разом з кінетичними даними, дають підстави стверджувати, що в умовах поліконденсації за реакцією Стегліха, особливо до конверсії 60-70%, процес реалізується через проміжну стадію утворення ангідриду.
В спектрах С13 ЯМР, крім сигналів ангідридних груп, були ідентифіковані також сигнали кінцевих груп, які утворюються при побічних перетвореннях активованої форми карбоксильної групи (схеми 3 і 4). Це означає, що ці процеси є конкурентними з основними не лише на глибоких стадіях, але очевидно відбуваються, хоча і менш виражено, на ранніх стадіях реакції.
Оптимізація умов проведення реакції утворення кополіестерів показала, що більш високі ступені поліконденсації досягаються в наступних умовах: і) допустимого надлишку діолу; іі) температурному діапазоні 286÷291К ііі) при використанні нуклеофільного агенту - 4-диметиламінопіридину (DMAP). Встановлено, що DMAP є найбільш ефективним нуклеофільним агентом і його використання дозволило отримати кополіестери зі ступенем поліконденсації у 2÷3 рази вищим у порівнянні з триетиламіном або діазабіциклооктаном.
Синтез потрійних кополіестерів двоосновних α-амінокислот з статистичним та регулярним розташуванням фрагментів у макромолекулі.
|
Схема 8. Будова потрійних кополіестерів |
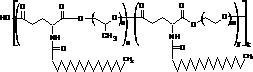
Третій розділ роботи присвячений дослідженням особливостей синтезу потрійних кополіестерів (схема 8). Видно (схема 8), що у склад макромолекул потрійних кополіестерів включено фрагменти двох поліетердіолів різної природи. Це надає можливість більш гнучко регулювати поверхнево-активні властивості макромолекул кополіестерів.
| |
Рис. 6. ПМР спектри потрійних кополіетерестерів Glu(St), DPG з, б) PEG-300, в) PEG-600 г) PEG-1000, д) PEG-2000 в хлороформі в порівнянні з кополіестеретером ко-Glu(St)-ко-DPG-ко-DEG а) |
В розділі розглянуто два основних шляхи одержання АПЕ такої будови: і) кополіконденсація N-заміщених Д-a-АК за реакцією Стегліха при співзавантаженні в реакційну суміш двох різних поліетердіолів; іі) синтез кополіестерів, цим же методом, але з наперед синтезованих форполімерів поліестеретерної природи. Останній шлях, крім введення в склад макромолекули різних за природою фрагментів поліетердіолів, дозволяє також вводити Д-a-АК з різними замісниками в N-положенні. При проведенні поліконденсації за першим методом, з одночасним використанням двох різних поліетердіолів в реакційній суміші постає питання про рівномірність розподілу ланок комономерів у складі макромолекули. Це особливо важливо при використанні поліетердіолів різної природи і різної молекулярної маси. Середньочисельну молекулярну масу потрійних кополіестерів оцінювали за даними GPS (табл. 2), а вміст кожного з комономерів встановлювали за допомогою ПМР спектроскопії. Для прикладу, на рис. 6 наведені ПМР спектри кополіестерів, які одержані при кополіконденсації Glu(St) з сумішшю DPG та різними за молекулярними масами PEG. Інтенсивність сигналу I (d=3,75 м. д) від протонів фрагментів PEG в порівнянні з інтегралом сигналу C (d=1,3 м. д) від протонів метиліденових груп алкільного ланцюга дозволяє розрахувати вміст фрагментів PEG і PPG у складі макромолекул.
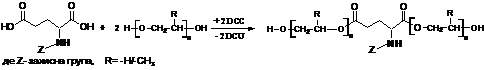 |
Встановлено, що особливості перебігу реакції при сумісному завантаженні реагентів повністю відповідають закономірностям поліконденсації за реакцією Стегліха які розглянуті раніше. Важливо, що використання двоосновних a-амінокислот з об’ємними замісниками захисту аміногрупи і поліетердіолів з молекулярною масою до 1000, дозволяє синтезувати кополіестеретери з ступенем кополіконденсації, які задовільно співпадають з розрахованими за рівнянням Карозерса.
Схема 9. Синтез диестерів за реакцією Стегліха при взаємодії N-похідних дикарбоновох a-амінокислот Glu(A) при надлишку поліетердіолу
За цим методом, синтезовані також потрійні кополіестеретери при використанні Д-a-АК з лабільними N-замісниками та входженням в макромолекулу різних за природою та молекулярною масою фрагментів поліетердіолів, або двох різних Д-a-АК при одному фрагменті PEG. В цьому випадку, проте, спостерігається нижча ніж прогнозована ступінь поліконденсації.
|
|
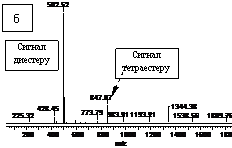
При використанні PEG з молекулярною масою більше 1000, у суміші з PPG з молекулярною масою до 300÷400, спостерігається пониження ступеня кополіконденсації та деяка перевага входження (на 15÷25%) поліетердіолу з меншою молекулярною масою. З цього можна зробити висновок, що в першу чергу, в реакційній масі вичерпується поліетердіол з меншою молекулярною масою. Крім того, це дає
Рис.7. Мас спектри зразків диестеретерів одержаних на основі Glu(Boc) і DEG при співвідношенні 1:5 а) і Glu(Boc) і DPG з співвідношенням 1:10 б).
підстави вважати, що при такому методі синтезу, поліоксиетиленові фрагменти різної природи нерівномірно розподілені вздовж макромолекули потрійного кополіестеретеру. Незважаючи на це, поліконденсацію за реакцією Стегліха, з деякими обумовленими обмеженнями, слід вважати зручним методом синтезу потрійних кополіестерів на основі N-похідних двоосновних a-амінокислот. Нерівномірний розподіл фрагментів поліетердіолів у макромолекулі було усунуто при проведенні синтезу потрійних поліестеретерів з наперед одержаних форполімерів (блоків). Крім того, при необхідності одержати кополіестеретери з лабільними захисними групами аміногрупи, з більшою ступеню поліконденсації ніж це дозволяє прямий синтез, проблема була вирішена також через застосування форполімерів.
Для здійснення такого методу синтезу кополіестерів була розроблена методика одержання форполімерів з кінцевими карбоксильними та кінцевими гідроксильними групами на основі N-заміщених Д-a-АК та поліетердіолів.
Синтез блоків з кінцевими гіроксильними групами було реалізовано через реакцію Стегліха (схема 9). Використання поліетердіолу в значному надлишку забезпечило пригнічення перебігу побічних реакцій за схемами 3 та 4, а також утворення поліестеретерів циклічної будови. В роботі наведено мас-спектроскопічне дослідження по оптимізації одержання блоків з кінцевими гідроксильними групами, яка зводилась до визначення мінімально необхідного надлишку поліоксиетердіолу для ефективного пригнічення реакції поліконденсації і побічних процесів. В результаті цього показано, що використання 7÷10 кратного мольного надлишку поліетердіолу у всіх випадках приводить до одержання блоку необхідної структури з виходом 80÷92 % в реакційній суміші і 60÷83% після очистки, в залежності від природи N-замісника і природи та молекулярної маси діолу. З наведених даних (рис. 7а) можна побачити, що мольне співвідношення 1:5 ще не повністю пригнічує перебіг реакції поліконденсації (спостерігається сигнал від тетраполіестеретеру), а також що спостерігаються сигнали від продуктів непродуктивного перегрупування активованої карбоксилльної групи.
| б | |||||
Рис. 8 ПМР спектри блоків (диестерів) з кінцевими групами синтезованих на основі солянокислої GluНCl та DEG. (а) і Glu(St) та DPG б). |

При співвідношенні 1:10 (рис. 7б) сигнал від диестеретеру є домінуючим. Сигнал тетраестеру у спектрі ідентифікується, але його кількість є незначною. А сигнали від продуктів перегрупування уже практично відсутні. Одержати блоки з кінцевими карбоксильними групами за реакцією Стегліха, як це було здійснено з блоками з кінцевими гідроксильними групами, неможливо.
Це повязано з тим, що надлишок карбоксильних груп, без сумніву не допускає протікання реакції поліконденсації, але імовірність процесів перегрупування та циклізації активованої карбоксильної групи в цьому випадку значно зростають. Тому для одержання блоків з кінцевими карбоксильними групами була використана реакція Бреннера (схема 10).
 Схема 10. Взаємодія N-похідних глутамінової кислоти з поліетердіолами за реакцією Бреннера.
Схема 10. Взаємодія N-похідних глутамінової кислоти з поліетердіолами за реакцією Бреннера.
Дана методика полягає у дії хлористим тіонілом на суміш N-похідної Д-a-АК та поліетердіолу в інертному розчиннику в присутності розкислювача.
Цікавим є той факт, що крім раніше зазначених N-похідних, в даній реакції можна використовувати Д-a-АК у формі її гідрохлориду, яка забезпечує неможливість перебігу рекції ацилування аміногрупи.
В роботі нами ця реакція першочергово розглядалась, як альтернатива реак ції Стегліха для проведення поліконденсації Д-a-АК з поліетердіолами в м’яких умовах, так як в літературі факти такої незворотньої поліконденсації відомі.
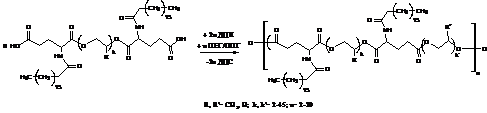
Схема 11. Хімізм утворення потрійного кополіестеру за участю діестеру через реакцію Стегліха.
Але у випадку Д-a-АК, в усіх співвідношеннях реагентів та температурах 273÷343K, а також з використанням в якості добавки диметилфораміду одержати кополіестеретери не вдалося. В умовах, які найбільше наближались до умов кополіконденсації, а також при підвищених температурах, спостерігалось суттєве осмолення реакційної маси. При цьому, єдиним продуктом, який вдається виділити є диестеретер з кінцевими вільними карбоксильними группами – продукт повного приєднання Д-a-АК по гідроксильних группах діолу (схема 10). Ідентифікація одержаних продуктів проводилась ПМР та ІЧ спектроскопією, вміст карбоксильних груп в зразках визначали потенціометричним та кондуктометричним титруванням. Для прикладу характерні ПМР-спектри диестеретерів одержаних на основі Glu(HCl) з DEG і Glu(St) з DPG наведені на рис. 8.
Таблиця 2.
Основні характеристики потрійних кополіестерів на основі попередньо синтезованих діестерів
Діестер | Діол | Співвідношення реагентів | Темература, °C | Вихід ДЦС,% | ММ, визначена за GPC, г/моль | Se | Sm | ||||
Glu(A) | Діол | ДЦК | Каталізатор | ||||||||
1 | Glu(St)-DPG-Glu(St) | PEG-400 | 8 | 9 | 16,8 | 1 | 15 | 98,8 | 8970 | 13,3 | 16,9 |
2 | Glu(St)-PPG400-Glu(St) | PEG-400 | 9 | 10 | 18,9 | 1,125 | 15 | 98,5 | 11630 | 14,4 | 20,1 |
3 | Glu(St)-PPG400-Glu(St) | PEG-300 | 9 | 10 | 16,8 | 1,125 | 15 | 97,7 | 9450 | 12,6 | 18,2 |
4 | Glu(St)-PPG1000-Glu(St) | PEG-400 | 8 | 9 | 6,8 | 1 | 5 | 7,8 | 10300 | 2,7 | 19,0 |
Таким чином, реакція Бреннера, в межах даного дослідження, виявилась вдалим інструментом для одержання блоків з рівноцінними кінцевими карбоксильними групами. В роботі наведено дані по оптимізації виходу диестерів синтезованих на основі солянокислої Glu та Glu(St) та поліетердіолів різної природи та молекулярної маси. Зокрема показано, що в оптимальних умовах вихід становить від 85% до 92% якщо використовували поліетердіоли з молекулярною масою не вище 1000. Використання діолів з молекулярною масою вище 1500 приводить до суттєвого пониження виходу. Крім того, одержати задовільні виходи диестерів з використанням в якості діола етиленгліколю та пропіленгліколю не вдалося, через протікання реації внутрішньомолекулярної циклізації.

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 |
