

Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Заключение
Электронное строение нитрозооксидов не может быть корректно описано только в рамках представлений о двухэлектронной природе химической связи и октетной теории Льюиса и Лэнгмюра. Теория резонанса Полинга дает более адекватное описание электронного распределения в нитрозооксидах в виде суперпозиции резонансных структур (схема 1), причем в наборе обязательно присутствуют резонансные состояния бирадикальной природы, например, структура III на схеме 1. Очевидно, что доля резонанса III может существенно изменяться в зависимости от особенностей строения исследуемого соединения. Вклад резонанса III может быть пренебрежимо мал для устойчивых состояний. Напротив, переходное состояние цис-транс-изомеризации ArNOO, в котором нарушается сопряжение в трехцентровой 4-π-системе электронов нитрозооксидной группы, a priori должно быть в значительной степени бирадикальным. Аналогичная ситуация возникает при вращении непредельных соединений вокруг двойной C=C связи. При данных обстоятельствах DFT-моделирование свойств нитрозооксидов необходимо проводить с учетом возможно бирадикальной природы ArNOO, проявляющейся в виде спиновой поляризации электронной плотности. В свою очередь, это означает, что конструирование волновой функции молекулы следует осуществлять с учетом возможной неэквивалентности электронного распределения, создаваемого электронами с противоположными спинами, т. е. необходимо использование неограниченного варианта Кона-Шама в DFT-расчетах.
Действительно, мы наблюдаем, что ограниченный формализм Кона-Шама (restricted Kohn-Sham, RKS) в ряде случаев приводит к нестабильности волновой функции изомеров ArNOO или переходных состояний. Однако, использование UKS (unrestricted Kohn-Sham) метода порождает другую проблему. Популярные функционалы плотности, в которых в обменный функционал добавляется хартри-фоковская обменная энергия, т. е. гибридные функционалы завышают степень бирадикальности синглетных состояний и их стабильность при использовании UKS-формализма. Данный недостаток проявляется и в расчетах нитрозооксидов. Видно (схема 5), что B3LYP функционал предсказывает заметную спиновую поляризацию даже в устойчивой структуре транс-PhNOO. Во всех примерах, приведенных на схеме 5, спиновая поляризация проявляется отчетливее в B3LYP-расчетах по сравнению с meta-GGA функционалом M06-L. Как результат, энтальпия переходного состояния конформационного превращения в HNOO, а также ΔH≠ цис-транс-изомеризации PhNOO существенно занижены в B3LYP расчетах вследствие переоценки стабильности этих переходных состояний (табл. 2 и 3). Характерно, что волновая функция переходного состояния син-анти конформационного перехода стабильна в рамках всех использованных функционалов. При этом результаты расчета с использованием B3LYP функционала хорошо согласуются с результатами других методов. Этот факт согласуется с изложенными выше представлениями о ненадежности оценок с помощью гибридных методов, если волновая функция исследуемого соединения имеет заметный вклад бирадикального резонанса.
Схема 5. Распределение спиновой плотности ( ‑ спиновая поляризация) в переходных состояниях конформационных переходов в HNOO (а) и PhNOO (с), а также в транс-форме PhNOO (б). Расчет методами B3LYP/6-311+G(d, p) (выделено курсивом) и M06‑L/6‑311+G(d, p).
|
|
|
а | б | с |
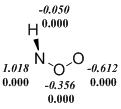
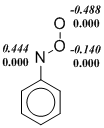
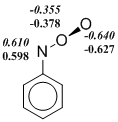
Таким образом, в настоящей работе проведено тестирование DFT функционалов с целью выбора оптимального теоретического метода для исследования строения, спектральных и энергетических свойств нитрозооксидов. Данные три критерия были проверены на пероксинитрене и фенилнитрозооксиде, выбранных в качестве модельных соединений. Включение в «чистые» функционалы плотности определенной доли хартри-фоковской обменной энергии существенно занижает значения активационных барьеров цис-транс-конформационного перехода как в HNOO, так и в PhNOO.
Выводы
1. Показано, что для исследования свойств нитрозооксидов не следует использовать DFT методы, включающие в функционал плотности хартри-фоковскую обменную энергию.
2. Изучение свойств ароматических нитрозооксидов целесообразно проводить с использованием функционалов плотности M06‑L, mPWPW91, OLYP и HCTH, которые наиболее надежно описывают строение, спектральные и энергетические свойства простейшего нитрозооксида – пероксинитрена и дают адекватные результаты для PhNOO в сравнении с высокоуровневыми расчетами.
Литература
THE STRUCTURE, ENERGY, AND SPECTRAL PROPERTIES STUDY OF NITROSO OXIDES BY DENSITY FUNCTIONAL THEORY
© Yusupova1+ Alfia Ravilevna, Safiullin Rustam2 Lutfullovich, Khursan1* Sergey Leonidovich
1Laboratory of Chemical Physics. Ufa Institute of Chemistry of the Russian Academy of Sciences.
71 pr. Oktyabrya, Ufa 450054, Russian Federation
E-mail: *****@***ru
2Laboratory of Chemical Kinetics. Ufa Institute of Chemistry of the Russian Academy of Sciences.
71 pr. Oktyabrya, Ufa 450054, Russian Federation
E-mail: *****@***ru
Keywords: nitroso oxides, trans-cis-isomerization, ortho-cyclization, density functional theory
Abstract
The choice of density functional for the study of the structure, spectral and energy properties of nitroso oxides – highly reactive particles with three-centered 4π-electron orbital system – was performed. It was shown that the hybrid DFT-methods overestimate considerably stability of singlet biradical states. Thus, an application of these methods for the wave function calculation was prevented in the case of nitroso oxides due to the multi-configurational character of Y. In contrast, generalized gradient approximations – M06‑L, mPWPW91, OLYP and HCTH showed good applicability to the calculation of the properties of nitroso oxides.
Introduction
Nitroso oxides RNOO are recognized to be highly reactive intermediates of photooxidation of HN3 and organic azides . The simplest representative of these compounds is peroxynitrene HNOO (X = HN, Scheme 1) is an isoelectronic analogue of ozone (X = O) and peroxymethylene H2COO (X = H2C). The functional group (XOO) of these compounds has three-centered 4π-electron orbital system. The electronic structure of nitroso oxides can be represented by a superposition of several resonance structures. The most important ones are shown in Scheme 1.
Scheme 1. Key resonance structures in 1,3-peroside dipole
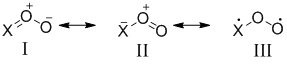
Nitroso oxides exist as two planar isomeric forms, cis and trans, which differ in the position of the terminal oxygen atom with respect to the R moiety :
Schem 2. The stable isomers of nitroso oxides.
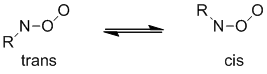
The correct description of systems with complex electronic structure inherent to nitroso oxides is a non-trivial quantum chemical problem. The wave function of nitroso oxides has multi-configuration nature. This fact limits the range of methods that adequately describe the system of interest.
The chemical properties of nitroso oxides have been mostly studied for aromatic nitroso oxides that have moderate reactivity due to the molecule stabilization through conjugation of the 4π‑electron system of ‑NOO with the aromatic moiety.
The structure of nitroso oxides was studied using various quantum-chemical methods, but calculations that include the major fraction of both static and dynamic electron correlation have only been performed for the simplest nitroso oxide representative, viz., HNOO . Theoretical calculations of the properties of aromatic nitroso oxides present challenge due to the complicated electronic structure of ArNOO. Calculations by multi-configurational methods would consume unreasonable computational and time resources. The use of the methods based on the DFT provides a good alternative. Theoretical simulation of the properties of aromatic nitroso oxides was performed most commonly using the B3LYP density functional. In some cases, the G3MP2B3 composite method was used where the calculation scheme also included optimization and calculation of frequencies at the B3LYP/6-31G(d) level of theory. However, it was noted that the use of the B3LYP functional for ArNOO, which is characterized by a noticeable contribution of biradical resonance III to the wave function (Scheme 1), results in a considerable overestimation of the stability of singlet states. It was shown that hybrid functionals containing a constant fraction of the Hartree−Fock exchange poorly describe the non-Lewis systems. On the other hand, mGGA gradient-corrected methods that account for electron density nonuniformity by inclusion of both density gradient and its Laplacian into the functional are characterized by the smallest deviation of the computational results from the experimental data or data obtained by the high-precision multi-configuration methods .
There are a large number of density functional methods at present. Nevertheless, there is no universal method that can properly describe the whole range of properties of a wide variety of chemical systems. In this regard, the purpose of this paper is to choose the method of density functional theory adequately describing the structure, spectral and energy properties of nitroso oxides.
Experimental
Selecting the calculation methods, we used the agreement between theoretical and experimental data as the main criterion. IR and UV spectroscopies are the main tools for experimental studies of nitroso oxides, so comparison of spectral characteristics is of interest. We used trans-HNOO as a model compound, since its experimental IR spectrum is known . In the paper cited, the structure of peroxynitrene and the IR spectrum of trans-HNOO were obtained by the CCSDT-3(Qf)/cc-pVTZ method that shows good agreement with experimental data. It allows us using it as the reference method for description of the structural features of nitroso oxides. Furthermore, because of the multi-configuration character of the HNOO wave function, we used for comparison the following approximations: MR-AQCC(8;7)/cc-pVTZ, CCSD(T)/6-311+G(d, p), as well as CASSCF (18;13), which includes all valence electrons . Correct reproduction of bond lengths is highly important in simulations of IR spectra. Therefore, when choosing the best DFT functional, we paid special attention to the conformity of N−O and O−O interatomic distances in the nitroso oxide group to the reference methods. Separate description of the movement of electrons with opposite spins is more flexible for building the wave function. Therefore, taking into account the partially biradical nature of nitroso oxides, the unrestricted Kohn−Sham method was used in all cases. The wave functions were constructed using the Dunning or Pople polarization basis sets of triple valent splitting−cc-pVTZ or 6-311+G(d, p). Quantum-chemical calculations were performed on a cluster super-computer of Ufa Institute of Chemistry of the RAS using revisionC01 , FireflyV.7.1 and CFOURV1.2 software packages. Visualization was performed in ChemCraft program .

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 |
