

Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Table 2. Reaction enthalpies (ΔH°) and activation enthalpies (ΔH≠) for trans-cis isomerization of peroxynitrene (kJ/mol)
Method | ∆H° | ∆H¹ | ∆H° | ∆H¹ | |
cc‑pVTZ | 6-311+G(d, p) | ||||
MR‑CISD(18;13)+Q [15] | ‑9.5 | 109.9 | ‑9.0 | 106.8 | |
CCSD(T) | – | – | ‑12.2 | – | |
M06‑L | ‑14.4 | 106.5 | ‑12.7 | 108.4 | |
mPWPW91 | ‑14.3 | 109.5 | ‑11.3 | 108.8 | |
OLYP | ‑13.9 | 114.7 | ‑10.2 | 114.1 | |
HCTH | ‑13.8 | 110.8 | ‑10.3 | 112.9 | |
B3LYP | – | – | ‑16.9 | 67.9 |
∆H° = H°cis – H°trans; ∆H¹ = H°TS – H°trans.
Overall, good agreement is observed between the results of the DFT methods chosen and those of the reference method. All the approximations used correctly predict the higher stability of the cis-isomer of peroxynitrene. In fact, if the Pople basis set is used, the stability of cis-HNOO in comparison with the trans-isomer is overestimated by only 1 kJ/mol (OLYP, HCTH) and the conformation barrier is overestimated by 2 kJ/mol (M06-L, mPWPW91). For comparison, Tables 1 and 2 show the results of calculations using the popular B3LYP functional. One can see that this method agrees with the reference method to within 0.01 Å as concerns bond lengths. However, it poorly reproduces the vibrational spectrum of HNOO and considerably underestimates (by 40 kJ/mol) the activation barrier ΔН≠ of the conformational transition of trans-HNOO to the cis-isomer.
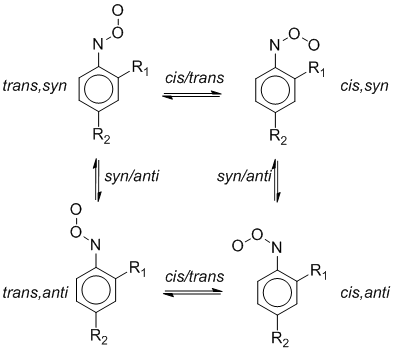
The conformational potential of ArNOO is determined by the availability of two rotation axes in the nitroso oxide group. Apart from cis and trans isomerization, aromatic nitroso oxides can undergo conformational transitions due to internal rotation of the molecule around the C−N nitroso oxide bond. The possible conformational transitions of arylnitroso oxides are shown in Scheme 4. According to Scheme 4, description of the conformational behavior of ortho-ArNOO (or meta-) requires calculations for four stable isomers and four transition states that connect the isomers on the conformational potential energy surface (PES). Effect of substituent, its position relative to nitroso oxide group and solvent effect on conformational transformations ArNOO were investigated in detail in . The present work deals with the conformational transformations of unsubstituted PhNOO (R1 = R2 = H, Scheme 4), which requires the calculation of only two isomers and three transition states.
Scheme 4. Conformational transformations in aromatic nitroso oxides
The results of the enthalpy and activation energy calculation for cis-trans isomerization in PhNOO are shown in Table 3. The M06-L and mPWPW91 approximations predict the thermodynamic preference of cis-isomer in the gas phase, while OLYP and HCTH functionals show similar stability of both conformers. The calculation of energy and structure using more sophisticated method CCSD(T) both by itself and in combination with DFT functionals shows again a slight difference in the energies of cis‑ and trans‑ isomers of PhNOO. The cis-trans ‑isomerization is accompanied by violation of π‑conjugation in nitroso oxide group and required overcoming the significant activation barrier ‑ about 60 ‑ 80 kJ/mol. This result explains the experimental observation that the cis‑ and trans‑ isomers of ArNOO behave as chemically independent species at room temperature and below, i. e. interconversion of isomers proceeds slowly enough in the ArNOO life time scale. Furthermore, the isomerisation into cis-form is the limiting step of irreversible consumption of trans‑ArNOO, therefore experimentally determined activation energy of trans‑ArNOO corresponds to that of the trans-cis-transition. The conformational barrier of the syn-anti transitions in arylnitroso oxides (ca. 20 ‑ 30 kJ/mol) appears due to conjugation of the four‑electron system of the nitroso oxide group with the aromatic π‑system. For example, in nitrosomethane where this kind of conjugation absent, the rotation barrier of the N=O group is ca. 4 kJ/mol . Table 3 presents the activation energies for both of the possible syn‑anti‑transitions in PhNOO: between cis ‑ and trans ‑isomers. Obviously, syn‑ and anti‑forms are mutually identical in PhNOO and the reaction enthalpy of cis‑cis‑ and trans‑trans‑isomerisations is equal to zero. The hybrid functional B3LYP, as well as in the case of peroxynitrene, significantly lowers the activation energy of cis‑trans‑transition in PhNOO (Table 3), while conformational barrier of syn‑anti‑transitions is reproduced more reliably.
Table 3. The reaction enthalpy ∆H and activation enthalpy ∆H (kJ/mol) of conformational transformation in PhNOO
Method | ∆H° | ∆H¹trans-cis | ∆H¹cis-cis | ∆H¹trans-trans |
gas phase | ||||
CCSD(T)/6-311+G(d, p) | -1.9 | ‑ | ‑ | ‑ |
CCSD(T)/M06-L/ | 1.9 | 79.6 | 16.9 | 29.4 |
CCSD(T)/mPWPW91/ | 2.5 | 82.1 | 24.3 | 23.5 |
CCSD(T)/OLYP/ | 2.3 | 83.0 | 16.9 | 23.3 |
CCSD(T)/HCTH/ | 1.6 | 81.0 | 17.3 | 23.3 |
CCSD(T)/B3LYP/ | 3.5 | 80.7 | 17.1 | 25.6 |
M06-L/ | -6.2 | 59.7 | 33.8 | 38.7 |
mPWPW91/ | -4.9 | 70.3 | 32.4 | 34.5 |
OLYP/ | 1.1 | 73.4 | 27.7 | 32.3 |
HCTH/ | 0.4 | 74.5 | 28.4 | 32.9 |
B3LYP/ | 3.6 | 48.3 | 34.6 | 42.7 |
acetonitrile | ||||
Experiment [1] | ‑ | 62 ± 2 | ||
M06-L/ | 0.1 | 67.6 | 36.5 | 45.0 |
mPWPW91/ | 1.2 | 77.1 | 34.7 | 40.6 |
OLYP/ | 7.1 | 80.2 | 30.1 | 38.3 |
HCTH/ | 6.4 | 81.3 | 30.7 | 38.9 |
The modeling of cis‑trans‑conformational transition in PhNOO including solvation was carried out to test the reliability of description of aromatic nitroso oxides properties using our chosen methods (Table 3). Effect of the solvent, namely acetonitrile, is accounted for using IEFPCM polarized continuum model. The PhNOO polarization is favored by polar solvent and, consequently, the latter increases the contribution of zwitterionic resonance structures (Scheme 1) into the wave function of the molecule. Trans‑form becomes more thermodynamically stable due to its more effective solvent stabilization in comparison with cis‑PhNOO. Activation energies of conformational transformation are in good agreement with experiment. The best agreement with experiment demonstrates the M06‑L functional.
Conclusion
Electronic structure of nitroso oxides can’t be correctly described only in the framework of the concept of two-electron nature of chemical bond and octet theory of Lewis and Langmuir. Pauling's resonance theory provides a more adequate description of the electron distribution in the nitroso oxides as a superposition of resonance structures (Scheme 1). It is noteworthy that the resonance states of radical nature should present necessarily in the set, like the structure III in Scheme 1. Obviously, the contribution of resonance III can vary substantially depending on the structural features of studied compound. It may be negligible for the stable states. On the contrary, the transition state of cis‑trans‑isomerization of ArNOO, in which conjugation in the three-centered 4π-electron orbital system of nitroso oxide group is broken, must be a priori largely biradical. A similar situation is known to arise in the case of rotation of unsaturated compounds around double C=C bond. In these circumstances, DFT-modeling of nitroso oxides properties should be carried out by taking into account the possible biradical nature of ArNOO which manifests itself as spin polarization of the electron density. Turn, this means that construction of wave function of a molecule should allow the possible nonequivalence of electron distribution with opposite spins, i. e. it is necessary to use the unrestricted Kohn-Sham method in DFT-calculations.
Indeed, we observe that the restricted Kohn-Sham formalism leads in some cases to the instability of wave function of ArNOO isomers or transition states. However, the use of UKS (unrestricted Kohn-Sham) method creates another problem. Popular density functionals, in which the Hartree-Fock exchange energy is included, i. e. hybrid functionals overstate the degree of biradicality and stability for singlet states in the frame of UKS-formalism. This disadvantage manifests itself in the calculation of nitroso oxides. It can be seen (Scheme 5), that the B3LYP functional predicts a significant spin polarization even in the stable trans-PhNOO structure. In all the examples given in Figure 5, the spin polarization is more pronounced in the B3LYP-calculations in comparison with the M06-L meta‑GGA functional. As a result, the transition state enthalpy of the HNOO conformational transition, as well as the ΔH≠cis-trans value for the PhNOO isomerization are lowered markedly in B3LYP-calculations due to the overestimation of the stability of these transition states (Table. 2 and 3). Noteworthy, the transition state wave function of the syn-anti conformational transition in PhNOO is stable for all used functionals. At that, the results of calculation using the B3LYP functional agree well with that of other methods. This fact is consistent with the above discussion on unreliable estimations by hybrid methods if the wave function of studied compound has a significant сontribution of biradical resonance.
Scheme 5. The spin density distribution ( ‑ spin polarization) in the transition states of conformational transitions in HNOO (a) and PhNOO (c), and in trans-form of PhNOO (b). The calculation by B3LYP/6-311+G(d, p) (in italics) and M06‑L/6‑311+G(d, p) approximations.
|
|
|
a | b | c |
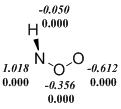
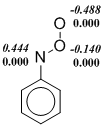
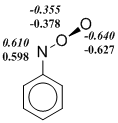
Thus, in this work the testing of DFT functionals was carried out to select the optimum theoretical method for studying the structural, spectral and energy properties of nitroso oxides. These three criteria are tested on peroxynitrene and phenilnitroso oxide chosen as model compounds. The inclusion of a certain percentage of the Hartree-Fock exchange energy in a "pure" density functional leads to significant underestimation of the activation enthalpy of cis-trans conformational transition both in HNOO and PhNOO.
Conclusion
3. It is shown that DFT methods which include the Hartree-Fock exchange energy should not be used to study the properties of nitroso oxides.
4. The study of the properties of aromatic nitroso oxides should be carried out using M06‑L, mPWPW91, OLYP and HCTH density functionals, that most reliably describe the structure, energy and spectral properties of the simplest nitroso oxide – peroxynitrene and give adequate results for PhNOO compared to the higher-level calculations.
References

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 |
