

Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
МИНИСТЕРСТВО ЗДРАВООХРАНЕНИЯ
РЕСПУБЛИКИ УЗБЕКИСТАН
Учебно – методический кабинет по Высшему и средне
Ташкентский фармацевтический институт
Факультет фармации
Кафедра токсикологической химии
Учебно– методическое указание и разработка к
лабораторному занятию по физико–химическим
методам анализа лекарственных средств
Метод высокоэффективной жидкостной хроматографии.
Применение этого метода анализа для определения качества некоторых лекарственных средств и остаточных количеств пестицидов в составе лекарственного растительного сырья
Ташкент - 2005
МИНИСТЕРСТВО ЗДРАВООХРАНЕНИЯ
РЕСПУБЛИКИ УЗБЕКИСТАН
Учебно - методический кабинет по Высшему и средне
медицинскому образованию
Ташкентский фармацевтический институт
«УТВЕРЖДАЮ»
Начальник Главного управления по кадрам, науке и учебным заведениям МЗ РУз _______________проф.
«___» ___________________2005 г.
Учебно–методическое указание и разработка предназначены студентам 4 курса для изучения одного из разделов предмета «Анализ качества лекарственных средств физико–химическими методами» «Метод высокоэффективной жидкостной хроматографии. Применение этого метода анализа для определения качества некоторых лекарственных средств и остаточных количеств пестицидов в составе лекарственного растительного сырья».
Составители: доценты , ,
Ассистенты ,
Под общей редакцией заведующего кафедрой токсикологической химии,
проф. .
Рецензенты: Директор vositalarini standartlash Ilmiy markazi», кандидат фармацевтических наук
,
Заведующий кафедрой аналитической химии Ташкентского фармацевтического института, кандидат химических наук, доцент
Настоящее учебно–методическое указание составлено исходя из требований существующей программы. Оно включает в себя следующее лабораторное занятие, рассчитанное для изучения хроматографического метода при высоких давлениях и применения его в анализе качества лекарственных средств и лекарственного растительного сырья.
Лабораторное занятие
Тема: «Метод высокоэффективной жидкостной хроматографии. Применение этого метода анализа для определения качества некоторых лекарственных средств и остаточных количеств пестицидов в составе лекарственного растительного сырья».
Учебная цель: Ознакомить студентов с методом высокоэффективной жидкостной хроматографии, хроматографическими терминами, подготовкой к хроматографическому анализу, подвижные фазы, условиями проведения анализов.
Продолжительность занятия: 6 часов.
Целевые задачи: проведение анализа лекарственных средств и лекарственного растительного сырья методом высокоэффективной жидкостной хроматографии.
ВЫСОКОЭФФЕКТИВНАЯ ЖИДКОСТНАЯ ХРОМАТОГРАФИЯ
Понятие высокоэффективной жидкостной хроматографии (ВЭЖХ)
Хроматография (от греческого слова chroma, родительный падеж chromatos - цвет, краска), физико-химический метод разделения и анализа смесей, основанный на распределении их компонентов между двумя фазами - неподвижной и подвижной (элюент), протекающей через неподвижную.
Хроматография широко применяется в лабораториях и в промышленности для качественного и количественного анализа многокомпонентных систем, контроля производства, особенно в связи с автоматизацией многих процессов, а также для препаративного (в том числе промышленного) выделения индивидуальных веществ (например, благородных металлов), разделения редких и рассеянных элементов.
В соответствии с агрегатным состоянием элюента различают газовую (ГХ, GC) и жидкостную хроматографию (ВЭЖХ, HPLC).
Высокоэффективная жидкостная хроматография (ВЭЖХ,) используется для анализа, разделения и очистки синтетических полимеров, лекарственных препаратов, детергентов, белков, гормонов и др. биологически важных соединений. Использование высокочувствительных детекторов позволяет работать с очень малыми количествами веществ (10-11-10-9 г), что исключительно важно в биологических и экологических исследованиях.
Метод ВЭЖХ осуществляется на различных жидкостных хроматографах. Современные жидкостные хроматографы предназначены для разделения сложных смесей веществ на отдельные компоненты и проведения качественного и количественного анализа компонентов разделяемой смеси.
Любой жидкостной хроматограф состоит из следующих базовых частей:
- узел подготовки элюента с емкостями для растворителей; насос; узел ввода пробы (инжектор); термостат колонок; хроматографическая колонка; детектор; регистратор (самописец, интегратор или компьютер); слив элюата или коллектор фракций.
Принцип действия хроматографа заключается в следующем: проба с помощью инжектора вводится в верхнюю часть хроматографической колонки. С помощью насоса анализируемая смесь прокачивается элюентом через хроматографическую колонку, в которой происходит разделение анализируемой смеси на отдельные вещества (компоненты). Вытекающий из колонки элюат, содержащий отдельные компоненты анализируемой смеси, детектируется детектором, показания которого регистрируются регистратором.
Наиболее часто встречающиеся в литературе термины ВЭЖХ
ЭЛЮЕНТ (подвижная фаза ПФ) - растворитель или смесь растворителей, предназначенная для прокачки анализируемой смеси через хроматографическую колонку.
АДСОРБЕНТ (НЕПОДВИЖНАЯ ФАЗА) - твердое вещество, способное удерживать растворенные вещества и используемые в хроматографии для заполнения хроматографических колонок.
ПРОБА – аликвота анализируемой смеси, вводимая в хроматограф.
СОРБАТ - компонент пробы, индивидуальное соединение.
НАГРУЗКА КОЛОНКИ – количество вещества в пробе. При введении в хроматограф большого объема пробы или концентрированной пробы может возникнуть перегрузка колонки, которая выражается расширением и искажением формы пика, а также зависимостью удерживания от вводимого количества вещества. На практике желательно работать при небольших нагрузках колонки.
ЭЛЮАТ – раствор, выходящий из хроматографической колонки.
ХРОМАТОГРАММА – графическое изображение результатов хроматографического процесса. Хроматограмма - кривая, описывающая зависимость концентрации анализируемых веществ в элюате от времени. Хроматограммой (с точки зрения аппаратурного оформления) можно назвать зависимость отклика детектора хроматографа от времени при прохождении элюата через ячейку детектора. Хроматограмма состоит из ряда пиков, каждый из которых при полном разделении соответствует одному компоненту анализируемой пробы. Площадь пика при небольшой нагрузке колонки прямо пропорциональна концентрации компонента в элюате.
ХРОМАТОГРАФИЧЕСКИЕ УСЛОВИЯ включают тип и марку используемого хроматографа, размеры хроматографической колонки и марку используемого в ней адсорбента, состав и расход элюента, тип элюирования (изократический или градиентный) и форму градиента, объем пробы, тип детектора и условия детектирования, температуру окружающей среды или термостата колонок.
ВРЕМЯ УДЕРЖИВАНИЯ ВЕЩЕСТВА (tR) - время пребывания адсорбата в хроматографе. На практике время удерживания определяют от момента ввода пробы вещества в хроматограф до момента регистрации максимальной амплитуды хроматографического пика адсорбата. Каждое вещество при одних и тех же хроматографических условиях имеет свое время удерживания. Это положение является основой идентификации (качественного анализа) компонентов разделяемой смеси по временам удерживания при жестком соблюдении постоянства условий эксперимента: состава элюента, расхода элюента (объемная скорость подачи элюента), использования одной и той же колонки, минимальных колебаниях температуры окружающей среды, сравнимых количеств вещества в хроматографическом пике анализируемой смеси и стандарта.
ВРЕМЯ УДЕРЖИВАНИЯ НЕСОРБИРУЕМОГО ВЕЩЕСТВА (tM) – время пребывания несорбируемого вещества в хроматографе. Определение времени удерживания несорбируемого вещества tM является важной проблемой, так как от точности определения tM зависит точность определения величины t'R, которая, в свою очередь, является исходной для расчета ряда хроматографических характеристик. Величина tM является характеристикой конкретной хроматографической системы при постоянном расходе элюента. Так как на практике не всегда удается подобрать полностью несорбируемый компонент, да и критерии подбора такого компонента весьма расплывчаты, на практике используют время удерживания наименее сорбируемого компонента, которое также обозначается tM
ПРИВЕДЕННОЕ ВРЕМЯ УДЕРЖИВАНИЯ (t'R) определяется по формуле: t'R = tR - tM
УДЕРЖИВАЕМЫЙ ОБЪЕМ (VR) компонента - объем элюента, вытекающий за время удерживания, определяется по формуле: VR = tR * w, где w - объемная скорость подачи элюента.
ОБЪЕМ УДЕРЖИВАНИЯ НЕСОРБИРУЕМОГО КОМПОНЕНТА (VM) – объем элюента, вытекающий за время пребывания несорбируемого компонента в хроматографе. VM включает в себя свободный объем колонки, объемы устройства ввода пробы и детектора, а также объемы коммуникаций между ними.
СВОБОДНЫЙ ОБЪЕМ КОЛОНКИ (V0) – часть объема колонки, не занятая сорбентом.
ПРИВЕДЕННЫЙ УДЕРЖИВАЕМЫЙ ОБЪЕМ ( V'R ) – определяется по формуле: V'R = VR - VM = t'R * w. Приведенный удерживаемый объем является объемом элюента, необходимым для вымывания анализируемого вещества только с поверхности адсорбента.
ФАКТОР ЕМКОСТИ (k') определяется по формуле k' = t'R / tM = V'R / VM. Фактор емкости является более универсальной, чем tR, характеристикой удерживания в том смысле, что k' не зависит от длины колонки и скорости подачи элюента.
ЭФФЕКТИВНОСТЬ (N) хроматографической колонки определяется экспериментально из хроматограммы по формуле: N = 5.545 * (tR / Wh )2, где:
N - эффективность хроматографической колонки в теоретических тарелках;
tR - время удерживания;
Wh - ширина пика на полувысоте.
Согласно формуле, эффективность колонки тем больше, чем уже хроматографический пик при тех же временах удерживания.
(ФАКТОР РАЗДЕЛЕНИЯ) – это способность хроматографической системы (адсорбента и подвижной фазы) делить данную пару соединений. В общем случае селективность является интегральным результатом межмолекулярных взаимодействий в хроматографической системе. Селективность хроматографической системы при разделении двух веществ определяется экспериментально по формуле a = t'R1/ t'R2
РАЗРЕШЕНИЕМ R<> R = 2 * ( tR1 - tR2)/( w1 + w2) . Для количественного анализа обычно достаточно разрешения R=1, так как в этом случае только примерно 2% площади пиков перекрываются. Разрешение двух хроматографических пиков зависит от селективности хроматографической системы, удерживания сорбатов и эффективности хроматографической колонки. Такая зависимость может быть выражена формулой: R = 1/4 * (a-1)/a * k'2/(k'2 + 1) * N1/2, где: k - фактор емкости второго хроматографического пика. Как следует из этой зависимости, разрешение в общем случае зависит от трех параметров – селективности a, удерживания k' и эффективности N; чем больше их значения, тем лучше разрешение.
Увеличение параметра N на практике означает замену имеющейся колонки на более эффективную. При этом следует помнить, что разрешение пропорционально квадратному корню из эффективности, то есть для улучшения разделения в два раза требуется колонка, в четыре раза более эффективная, чем данная.
Как правило, чем больше удерживание пары пиков (больше значение k'2), тем лучше она разделяется. Практически увеличения удерживания добиваются путем уменьшения концентрации полярной добавки или модификатора в элюенте.
Два упомянутых выше способа являются скорее "экстенсивными", и способны лишь отчасти улучшить разрешение. На практике они могут применяться, к примеру, при воспроизведении стандартных методик на местах.
Значительного увеличения разрешения можно достигнуть лишь путем регулирования селективности, a изменения селективности можно добиться, заменив неподвижную фазу или изменив состав элюента.
В процессе перемещения через разделительную колонку хроматографическая зона вещества размывается. Для характеристики интенсивности размывания используется величина Н – высота, эквивалентная теоретической тарелке (ВЭТТ). Качество хроматографической колонки характеризуется эффективностью N – величиной, обратной ВЭТТ: N = L / N, где L – длина колонки. Эффективность хроматографических колонок экспериментально определяется по известной формуле: N = 5.545 * (tR / w )2, где:
Термин "теоретическая тарелка" (Т. Т.) появился благодаря концепции, предложенной Мартином и Синджем. Хроматографическая колонка разбивается на ряд "тарелок" (подобно ректификационной колонне) – слоев, на каждом из которых происходит единичный акт адсорбции-десорбции при условии отсутствия диффузионного переноса анализируемого вещества между слоями.
Внешний вид хроматограммы: 
КЛАССИФИКАЦИЯ СОРБЕНТОВ ПРИМЕНЯЕМЫХ ПРИ ВЭЖХ АНАЛИЗОВ
Основными группами сорбентов являются:
1. Поверхностно - сорбенты, представляющие собой непроницаемое для растворителя твердое ядро из стекла, на поверхность которого занесен тонкий слой пористого абсорбента, обычно силикагеля;
2. Пористые сорбенты на основе силикагеля;
3. Пористые сорбенты на основе оксида алюминия;
4. Пористые сорбенты на полимерной основе.
Сорбенты первой группы были исторически первыми, стимулировавшими быстрый рост ВЭЖХ. Они представляют собой стеклянные микрошарики размером 35-50 мкм, на поверхности которых различными способами закрепляется слой силикагеля или оксида алюминия толщиной в 1-2 мкм. Этот слой может либо использоваться для разделения методом адсорбционной хроматографии, либо модифицироваться нанесением подвижной фазы.
Недостатки физически нанесенных фаз стимулировали быстрое развитие методов химической прививки фаз, далее широко использовавшихся для других групп сорбентов.
Дальнейшее быстрое развитие ВЭЖХ базировалось на новом поколении сорбентов: микрочастицах диаметром от 3 до 10 мкм, главным образом на основе силикагеля, частично оксида алюминия, а в последнее время на основе пористых полимеров.
СИЛИКАГЕЛЬ, ЕГО СТРУКТУРА И ХИМИЯ ПОВЕРХНОСТИ
Силикагель представляет собой почти чистый диоксид кремния SiО2, однако технические его сорта содержат примеси. Силикагель всегда содержит большие, или меньшие количества адсорбированной воды (этом основано его широкое использование в качестве осушителя). Силикагель имеет разную поверхность, составляющую обычно 100-600 м2 /г, и значительный объем пор (0,5-1,2 мл / ) с преобладанием пор диаметром от 5 до 15 нм.
Настоящее время разные фирмы производят более 200 сорбентов для ВЭЖХ на основе силикагеля. Эти сорбенты обладают хорошей механической прочностью, совместимостью с большинством растворителей, возможности получения частиц микронного размера, достижения высокой эффективности колонок и низкой каталитической активности диоксида кремния.
ПРИВИТЫЕ СОРБЕНТЫ НА ОСНОВЕ СИЛИКАГЕЛЯ ДЛЯ НОРМАЛЬНОЙ И ОБРАЩЕННОЙ ХРОМАТОГРАФИИ
Сорбенты с химически привитыми фазами на основе силикагеля появились позже сорбентов, на которые неподвижная фаза (виде индивидуальных веществ или, чаще, полимеров различной структуры и полярности) наносилась физически, аналогично тому, как фазу наносили и продолжают наносить в газожидкостной хроматографии.
Нанесенная фаза довольно быстро смывается растворителем ( быстрее, чем она испаряется или изменяется в газожидкостной хроматографии), параметры удерживания постоянно меняются, препаративно - собираемые фракции загрязняются фазой.
Использование растворителя, насыщенного неподвижной фазой, позволило несколько повысить стабильность таких сорбентов и колонок, однако большинство недостатков при этом осталось.
Можно отметить следующие преимущества, обеспечивающие преобладающее использование привитых сорбентов на основе силикагеля: механическая устойчивость к высоким давлениям; отсутствие перехода привитой фазы в растворитель в процессе хроматографического разделения ( не протекают реакции, приводящие к химическому отщеплению привитой фазы ); устойчивость к действию растворителей, температуры, воды, рН; быстрота установления равновесия при смене элюента, что обеспечивает оперативность работы и возможность работы в градиентном режиме с быстрым возвратом к исходному режиму; возможность варьировать в широких пределах селективность за счет изменения степени прививки, дополнительной химической обработки и замены растворителя.
Наиболее популярными являются так называемые обращенные привитые фазы, применяемые в обращенно - ВЭЖХ. Понятие пришло от классической ЖХ на силикагеле, где в системе подвижная фаза неполярная, а неподвижная полярная (гексан и силикагель ). По этому принципу система должна иметь полярную подвижную фазу и неполярную неподвижную ( водный метанол и октадецилсилан на силикагеле, привитый химически ). Названия обращенно-сорбент и обращенно-система не являются особенно удачными или понятными, однако так как это название общепринято, то мы будем его придерживаться.
Работа по улучшению качества привитого слоя велась разными учеными постоянно и в разных направлениях. С целью устранения полимеризации прививаемого агента было предложено использовать монохлорсиланы вместо трихлорсиланов как раньше использовали (диметилокта-децилхлорсилан, диметилоктилхлорсилан и др.), которые по природе своей монофункциональны и могут дать только мономерный привитой слой. Вода дезактивирует монохлорсиланы, вступая с ними в реакцию, однако в реакцию прививки они уже не вступают и после окончания реакции отмываются вместе не прореагировавшим исходным алкилдиметилхлорсиланом. С целью устранения влияния остаточных силанольных групп было предложено после проведения прививки вести так называемое « замещение », или « кэппинг ». В этом случае после прививки основной фазы сорбент обрабатывают сильными реагентами с минимальным мольным объемом ( пример, триметилхлорсиланом), которые блокируют основную массу не прореагировавших силанольных групп.
Силикагель, используемый как матрица для последующей прививки неподвижной фазы, играет важнейшую роль в определении конечных свойств получаемого сорбента. Он имеет пространственно - структуру, образованную диоксидом кремния в процессе образования золя, геля и последующей его сушки с удалением физически сорбированной воды. В зависимости от условий формования силикагеля могут быть получены образцы со средними размерами пор от 3 до 10 нм. За счет последующей гидротермальной обработки силикагеля может быть достигнуто значительное увеличение размера пор (20-50 нм и более ) при сохранении в основном объема пор. Методами формования микросферических сорбентов для ВЭЖХ из тетраэтоксисилана за счет варьирования условий формования и отвердения, выбора растворителей и т. удается добиться получения силикагеля с достаточно высокой пористостью ( объем пор 0,7-2 мл / ) и порами от 5 до 400 нм и более.
Кроме размера пор, большую роль играет объем пор силикагеля и его поверхность. Если рост поверхности дает увеличение количества силанольных групп ( плотность около 5 на 1 и 2 нм ) и количества привитой фазы при равной плотности прививки, то рост объема пор играет сложную роль. При увеличении объема пор не только увеличивается проницаемость силикагеля, но и уменьшается объем самого диоксида кремния и соответственно прочность силикагеля; он легче разрушается в процессе транспортировки, при набивке колонок, повышении давления при эксплуатации колонок. Правда, прочность определяется не только толщиной стенок ячеек силикагеля, но и их структурой.
Практически все фирмы выпускают основной ассортимент привитых фаз, который включает С18, С8, нитрильную и аминофазы, различающиеся также по зернения. Каждая конкретная колонка имеет при себе паспорт, в котором указывается ее параметры, такие как, тип и название сорбента, его зернение, верхняя и нижняя граница показателя рН при котором допускается провести хроматографические разделение, размеры колонки, максимальное рабочее давление и т. д.
ПОДВИЖНАЯ ФАЗА ДЛЯ ВЭЖХ
Роль подвижной фазы в жидкостной хроматографии весьма многообразна. Наряду с чисто транспортной функцией растворитель активно участвует в самом процессе разделения и оказывает существенное влияние на возможности детектирования. Часто незначительное изменение состава подвижной фазы дает возможность оптимизировать процесс, улучшить форму пиков, разрешение отдельных компонентов и даже изменить механизм разделения. Поэтому при выборе растворителей необходимо учитывать весь комплекс их свойств, в той или иной степени влияющих на проведение хроматографического эксперимента.
ОСНОВНЫЕ ТРЕБОВАНИЯ К РАСТВОРИТЕЛЯМ
Растворители, применяемые в ВЭЖХ, должны удовлетворять следующим основным требованиям: чистота, химическая инертность, совместимость с детектором, достаточная растворяющая способность по отношению к анализируемым веществам низкая вязкость, безопасность, доступность. В некоторых случаях существенное значение имеют смешиваемость с другими растворителями, температура кипения и возможность легкого извлечения вещества из элюата.
Чистота растворителя в жидкостной хроматографии имеет очень большое значение, так как различные примеси в подвижной фазе влияют на все основные стадии процесса: подачу растворителя, разделение в колонке, детектирование и воспроизводимость результатов. Требуемая степень чистоты растворителя определяется выбранным вариантом разделения и используемой аппаратурой.
Наличие примесей в растворителе может вызвать следующие типичные затруднения.
1 . Ухудшение эффективности разделения и воспроизводимости результатов (неконтролируемая влажность растворителя в адсорбционной хроматографии ).
2. Сильное отклонение нулевой линии и образование ложных пиков при градиентном элюировании.
3. Ухудшение возможностей детектирования ( олефинов в парафиновых углеводородах при УФ -, примесь этанола в хлороформе при ИК - ).
4. Порча сорбента: примеси оснований приводят к растворению силикагеля;
примеси диенов и других лабильных соединений осмоляются и блокируют поверхность адсорбентов, особенно оксида алюминия; примеси карбонильных соединений реагируют с привитыми сорбентами, содержащими аминогруппу; пероксиды окисляют привитые фазы и полистирольные гели.
5. Загрязнение веществ, выделяемых из элюата. В препаративной хроматографии приходится выделять вещества из очень разбавленных растворов. При этом даже незначительные примеси или добавки, которые не мешают аналитическому разделению, могут концентрироваться в извлекаемом веществе, существенно снижая его чистоту.
6. Разложение или химическое изменение компонентов пробы ( примеры многих металлоорганических соединений, окисление лабильных веществ пероксидами или растворенным кислородом ).
7. Коррозия аппаратуры ( HCl в хлорсодержащих растворителях ).
Химическая инертность. Все, что сказано выше о химически активных примесях имеет гораздо большее значение применительно к химической активности самих растворителей. Дополнительно можно отметить, что такие классы соединений, как кетоны, алифатические и ароматические амины, следует применять с особой осторожностью и только в тех случаях, когда их трудно заменить более стабильными растворителями. Такие элюенты, как хлорорганические соединения, тетрагидрофуран и другие простые эфиры, следует использовать только свежеочищенными.
Совместимость с детектором. Наиболее распространенными детекторами в настоящее время являются УФ - и дифференциальные рефрактометры.
Возможность использования тех или иных растворителей в сочетании с УФ - детектированием принято определять минимальной длиной волны, на которой при оптическом пути 10 мм падение интенсивности светового потока составляет 90%. При УФ - детектировании практически не могут быть использованы такие растворители, как бензол, толуол, тетрахлорид углерода, диметилформамид и хлороформ, а также сложные эфиры и кетоны из – за высокой светопоглощаемости при длинах волн в котором поглощают большинство веществ.
С рефрактометрическим детектором в принципе можно применять любые растворители, но его чувствительность определяется разностью показателей преломления растворителя и анализируемого вещества. Поэтому при выборе растворителе следует учитывать его показатель преломления.
Вязкость растворителя должна быть по возможности низкой, так как ее повышение ведет к ухудшению массопередачи, а тем самым и эффективности разделения, а также затрудняет работу насосов. При прочих равных условиях следует выбирать растворители, имеющие вязкость 0,5-0,7 мПа при температуре разделения.
Безопасность работы с теми или иными растворителями определяется их воспламеняемостью и токсичностью. Практически все растворители, применяемые в ВЭЖХ, либо имеют весьма низкую температуру вспышки, либо в определенной степени токсичны. Поэтому помещение, в котором проводят работы по жидкостной хроматографии, должно иметь эффективную проточною - вентиляцию. На рабочем месте недопустимы плохо продуваемые и застойные зоны, так как в них могут накапливаться пары растворителей, имеющие большую плотность чем воздух. Нижний предел взрываемости многих растворителей составляет 1-2%, поэтому в застойных зонах возможно образование взрывоопасной смеси.
Во всех случаях следует выбирать наименее пожароопасные и токсичные растворители, руководствуясь соответствующими данными, приведенных в справочниках. Так, диэтиловый эфир можно заменить диизопропиловым, а бензол толуолом практически без ущерба для разделения.
С точки зрения, безопасности токсичность является более важным фактором, чем пожароопасность. При хорошей организации рабочего места и тщательном соблюдении правил техники безопасности опасность загорания практически исключена, а контакта с растворителем полностью избежать невозможно. Многие ароматические и хлорсодержащие растворители обладают способностью накапливаться в организме человека. По последним данным, некоторые из них, считавшиеся ранее малотоксичными (тетрахлорэтилен) являются канцерогенами, поэтому работа с этими растворителями требует осторожности.
Температура кипения менее существенный фактор, чем характеристики, рассмотренные выше. Ее следует учитывать в основном в двух аспектах: в надежности работы насосов и детекторов и легкости выделения вещества из элюата.
Низкокипящие растворители часто образуют пузырьки в насосах и детекторах. При использовании наиболее распространенных в настоящее время плунжерных насосов вероятность образования пузырьков тем больше, чем выше давление паров растворителя и скорость плунжера в фазе всасывания. Наличие пузырьков в насосе резко снижает точность подачи растворителя, а пузырьки в детекторе вызывают сильный шум и нестабильность нулевой линии. Для предотвращения этого явления проще всего применять растворители, температура кипения которых по крайней мере на 20-50 °С выше комнатной. С другой стороны, при необходимости препаративного выделения вещества нецелесообразно использовать высококипящие растворители.
Смешиваемость с другими растворителями необходимо учитывать при работе в режиме градиентного элюирования и при подготовке анализируемого образца с использованием предварительного экстракционного разделения.
Следует помнить, что подвижная фаза в ВЭЖХ всегда должна быть гомогенной.
Однако такие важные полярные растворители, как метанол и ацетонитрил, ограниченно смешиваются с гексаном. Для расширения диапазона концентраций, соответствующих гомогенным смесям, гексан заменяют на циклогексан или изооктан. Полная смешиваемость в подобных системах достигается заменой полярного компонента на этанол или изопропанол.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ КОМПОНЕНТОВ ТАБЛЕТОК ЦИТРАМОНА
5 таблеток растирают в ступке до получения мелко дисперсного порошка. Из этого порошка берут навеску около 0,25 г (т. н.) и переносят в мерную колбу объемом 25 мл. Навеску растворяют в небольшом объеме подвижной фазы. Раствор взбалтывают в течение 3-х мин на водяной бане при температуре 600С. После этого раствор охлаждают до комнатной температуры и доводят объем до метки подвижной фазой. Раствор фильтруют через бумажный фильтр и первые 5 мл фильтрата отбрасывают. Из оставшегося фильтрата берут 1,0 мл и переносят в мерную колбу объемом 25 мл и раствор доводят до метки подвижной фазой. Раствор хорошо перемешивают и фильтруют через фильтр «Миллипор» (размер пор 0,45 мкм). 20 мкл профильтрованного раствора вводят в инжектор хроматографа и хроматографируют в следующих условиях:
колонка Zorbax Eclipse XDB C-8, размером 4,6x150 мм, размеры частиц 5 мкм;
подвижная фаза: очищенная вода 305 мл, метанол – 190 мл, ледяная уксусная кислота – 5мл, дегазированная любым удобным способом;
скорость подвижной фазы 1,0 мл/мин;
длина волны детектора – 272 нм.
Приготовление стандартных растворов:
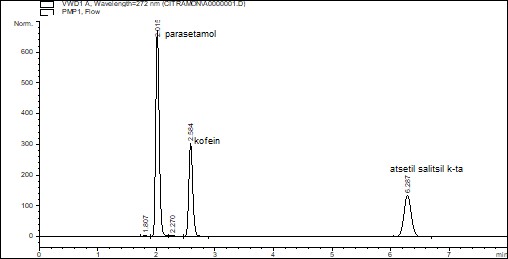
В мерную колбу с объемом 25 мл вносят отдельно стандартных образцов парацетамола, кофеина, ацетилсалициловой кислоты 0,04, 0,03, 0,044 г соответственно и растворяют их в небольших количествах подвижных фаз. Далее объем раствора доводят подвижной фазой до метки. Их этих растворов отбирают парацетамола 5,0 мл, ацетилсалициловой кислоты 5,0 мл, кофеина 1,0 мл и вносят в мерную колбу объемом 25 мл и объем доводят до метки подвижной фазой. Полученный раствор фильтруют через фильтр «Миллипор» размером пор 0,45 мкм и вводят в инжектор хроматографа. Из колонки компоненты таблеток «Цитрамона» выходят в следующем порядке: парацетамол, кофеин и ацетилсалициловая кислота.
Количественное содержание компонентов в таблетках «Цитрамон» вычисляют по следующей формуле:
![]()
![]()
где, S исп– площадь пика исследуемого вещества;
aст – вес стандартного образца;
Sст – площадь пика стандартного образца;
а - вес порошка растертой таблетки;
W - процентное содержание стандартных образцов;
b – средний вес таблеток;
V1 и V2 – объемы разведения стандартного и исследуемого образца.
Содержания ацетилсалициловой кислоты, парацетамола и кофеина должны быть 0,209-0,231, 0,19-0,21, 0,027-0,033 г/таб., соответственно.
ОПРЕДЕЛЕНИЕ ОСТАТОЧНОГО КОЛИЧЕСТВА ПЕСТИЦИДОВ В ЛЕКАРСТВЕННОМ РАСТИТЕЛЬНОМ СЫРЬЕ, С ЦЕЛЬЮ КОНТРОЛЯ КАЧЕСТВА СЫРЬЯ
Пробу растительного материала отбирают в соответствии с «Унифицированными правилами отбора проб для определения микроколичеств пестицидов в сельхозпродукции; продуктах питания и объектах окружающей среды». Растительную пробу измельчают до мелких частей и отбирают на анализ в количестве 10 г.
10 г измельченного растительного материала помещают в коническую колбу объемом 250 мл, заливают 50 мл ацетонитрила и взбалтывают в аппарате для встряхивания жидкостей в течении 30 мин. Органический слой количественно переносят в делительную воронку объемом 500 мл, фильтруя через бумажный фильтр. Навеску сырья ещё дважды обрабатывают с 30 мл ацетонитрила в течении 10 мин. Полученные экстракты объединяют, после чего для удаления балластных веществ обрабатывают петролейным эфиром 3 раза по 30, 20, 20 мл. Ацетонитрил отделяют от петролейного эфира и последний отбрасывают. К оставшемуся слою ацетонитрила добавляют 50 мл воды и 10 мл насыщенного раствора хлорида натрия. К смеси добавляют 50 мл гексана, энергично взбалтывают в течении 10 минут и оставляют для разделения фаз. Верхний гексановый слой переносят в круглодонную колбу объемом 250 мл, фильтруя через бумажный фильтр, содержащий 5 г безводного сульфата натрия, предварительно смоченного растворителем. Экстрагирование ацетонитрильного раствора с 50 мл н-гексана повторяют ещё 2 раза. Гексановые экстракты объединяют, упаривают органический растворитель на роторно-вакуумном испарителе при 400С до объема 1-2 мл. Оставшийся после отгона растворителя экстракт переносят в фарфоровую чашку, колбу промывают два раза с 1-2 мл гексана и присоединяют к первому экстракту. Объединенные экстракты упаривают досуха в токе воздуха. Сухой остаток растворяют в 1,0 мл ацетонитрила, фильтруют через фильтр «Миллипор» с размером пор 0,45 мкм и проводят хроматографический анализ на жидкостном хроматографе, снабженном градиентным насосом и дегазатором.
Приготовление растворов стандартного образца.
0,05 г данитола или 0,05 г циперметрина (точная навеска) переносят в мерную колбу объемом 50 мл и добавляют 3-5 мл ацетонитрила, растворяют пестициды, после чего доводят объем раствора до метки ацетонитрилом. 1 мл этого раствора содержит 0,001 г пестицида.
Приготовление 10 мМ раствора КН2РО4.
0,680 г КН2РО4 растворяют в очищенной воде, доводят объем раствора до 500 мл. Затем 25% раствором Н3РО4 рН этого раствора потенциометрически доводят до 3,0.
Условия хроматографирования.
В работе используется детектор типа UV/VIS с переменной длиной волны и колонку наполненную с обращенно – фазным сорбентом марки Zorbax Eclipse XDB C18, с размером частиц 3,5 мкм. Размер колонки 150х3 мм. Анализ проводят в линейном градиентном режиме со скоростью потока подвижной фазы 0,5 мл/мин и детектированием при 278 нм.
Раствор А – 10 мМ раствор КН2РО4 (рН=3);
Раствор Б – ацетонитрил.
Градиент: 0 мин: А – 80%, Б – 20%; 7-20 мин: А – 25%, Б – 75%; 24 мин А – 80%, Б – 20% .
Продолжительность анализа составляет 24 минут. В этих условиях время удерживания данитола составляет 17,2 и изомеров циперметрина составляет 18,5 и 19,1 минут. Предел обнаружения пестицидов данитола и циперметрина в этих условия составляет 1 мкг/мл.
В хроматограмме характерным для обнаружения циперметрина является наличие цис-, транс - изомеров в том соотношении, которое соответствует требованиям НТД. Появление двух пиков и значение соотношений цис-, транс - изомеров можно использовать как один из показателей, способствующих идентификации пестицида.
Количественное содержание пестицидов в лекарственном растительном сырье рассчитывают по формуле:
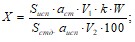
где,
к – коэффициент перерасчета, 1,39-для циперметрина, 1,28-для данитола
Sст – площадь пика стандартного раствора;
Sисп – площадь пика исследуемого вещества;
аст - навеска стандартного образца;
аисп – навеска исследуемого лекарственного растительного сырья;
V1 и V2 – объемы разведения стандартного и исследуемого образца.
ВОПРОСЫ ДЛЯ САМОСТОЯТЕЛЬНОЙ ПОДГОТОВКИ
Дайте характеристику методу ВЭЖХ. Объясните порядок и принцип работы хроматографа. Перечислите наиболее часто встречающие термины ВЭЖХ. Дайте определение терминам времена удерживания. Дайте определение терминам объемы удерживания. Дайте определение терминам элюат, адсорбент, сорбент, пробы, нагрузки колонки. Дайте определение терминам хроматограмма, хроматографические условия. Дайте определение терминам разделение, разрешение. Объясните порядок проведение анализа лекарственных средств. Объясните порядок проведение контроля качества лекарственного растительного сырья.
ЛИТЕРАТУРА
Государственная фармакопея ХI изд. М. Медицина. 1990 г. , , Брауде высокоэффективная жидкостная хроматография. –М. Химия. –1986. –206с. идкостная хроматография при высоких давлениях. Пер. с англ. М., Мир. –1980. –245 с. Методические указания “Определение циперметрина и данитола в лекарственном растительном сырье мяты перечной, календулы лекарственной, стевии ребо методом ВЭЖХ” Ташкент. -2004. -9 с.
