


Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Сосудистая терапия тадалафилом, ингибитором фосфодиэстеразы -5.
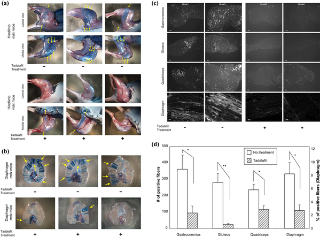
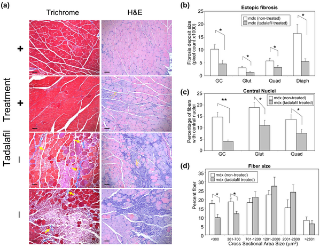
Для проверки гипотезы, что (I) предотвращение дегенерации миофибрилл может мешать уменьшать развитие мышечной дистрофии, и (II) агенты направлены на NO - цГМФ путь могут быть использованы в качестве терапевтических кандидатов, MDX мышам давали перорально тадалафил, ингибитор фосфодиэстеразы 5 ( PDE5I ). Этот препарат повышает внутриклеточный уровень цГМФ в гладкомышечных клетках сосудов, вызывает расширение кровеносных сосудов и увеличивает приток крови к органу-мишени. Без лечения MDX мыши показали повышенное количество поврежденных мышечных волокон в задних конечностях и диафрагме, как показал их синий цвет после инъекции красителя Эванса синего (рис. 7а, верхней панели 6 и 7б, верхняя 3 панели). Когда MDX мышей обрабатывали тадалафилом, количество умирающих волокон снижалась (фиг. 7а нижней панели 6 и 7б, нижняя панель 3 ) . Когда срезы мышц задних конечностей и диафрагмы мышей MDX оцениваются флуоресцентной микроскопией, MDX мыши без лечения показали повышенние красного флуоресцентного окрашивания то время как воздействие тадалафила снизило травму икроножной, ягодичной мышцы, четырехглавой мышцы и мышц диафрагмы ( рис. 7 и 7D) . Для дальнейшего подтверждения этого вывода использовалась обычная гистология с трехцветной окраской и гематоксилин-эозином (H & E) (рис. 8а). Обширные повреждения мышечных волокон и фиброз (рис. 8а, синий депозит в интерстициальной области в трехцветного окрашивания, желтые стрелки) занимает видное место у необработанных мышей MDX. Степень фиброза была снижена при лечении (рис. 8b, р <0,05) . Количество мышечных волокон с центральными ядрами сократилось при лечении (рис. 8в). Изменение размера волокон заметно у необработанных мышей MDX, было уменьшено при лечении тадалофилом (рис. 8d, р <0,05) . Эти данные предполагают, что защитный эффект на миофибриллы тадалафила может замедлить развитие болезни.
![]()
Обсуждение
Функциональная ишемия может стать существенной причиной мышечной дистрофии.
До сих пор нет единой точки зрения по поводу роли регулирования кровотока при МДД, изучения которой были начаты еще в середине 19-го века. Самый важный вопрос - играют ли оставшиеся нарушения кровообращения в скелетных мышцах основную роль в патогенезе МДД. С помощью вновь созданных анализов мы исследовали в естественных условиях гибель мышечных волокон, в сочетании с анализом микроциркуляции, мы зафиксировали доказательства того, что поддерживает важнейшую роль регуляции сосудистого тонуса в патофизиологии скелетных мышц у MDX мышей и показали, что лекарственные средства направленные на регуляцию сосудов эффективны для уменьшения повреждений мышечных волокон у этих мышей. Этот вывод согласуется с предыдущими сообщениями, что избыточная экспрессия трансгенов нОАС у мышей MDX [ 41 ], [42 ] или дистрофина в гладкомышечных клетках сосудов [43] уменьшают повреждения мышц. Исследования экспрессии нОАС у мышей MDX с использованием специфического промотора скелетных мышц [41], [42], и послужили основой для утверждения, что NО защищает мышечные волокна. Таким образом, предыдущие исследования не исключают возможность того, что NO производится из мышечных волокон, чтобы защитить мышечные волокна, действуя на сосудистую регуляцию. Прошлые отчеты подтвердили, что нОАС выражение в миофибриллах контролирует расширение кровеносных сосудов в скелетных мышцах [8]. Теория «Функциональной ишемии» мышечной дистрофии показала, что под напряжением сосудосуживающих механизмов мышц пациентов не в состоянии увеличить приток крови в норму даже после сокращения мышц [ 4], [ 5]. До сих пор одним из технических ограничений исследований гибели клеток было тог, что гибель клеток миофибрилл всегда увеличена в мышцах при мышечной дистрофии (см. фиг.7 алфавит и г). В обычных анализах существует значительное количество клеток даже перед фармакологическим/молекулярным вмешательством, маскирующих эффект вмешательства, даже если они вызывают дальнейшее повреждение клетки. Наш новый метод клеточной гибели мышечных волокон устраняет уже мертвые клетки с помощью мембранного потенциал - зависимого красителя, DiOC6 , который проникает только в живые клетки в начале эксперимента. Этот новый подход позволил количественно определить специфическое действие фармакологических вмешательств. С помощью непосредственного наблюдения за живыми животными нами было зафиксировано полное отсутствие стимулирования производства двух молекул сосудорасширяющих веществ, NO и H2O2 , в ответ на сокращение мышц: возможной причиной функциональной ишемии. Подробные наблюдения обнаружили слегка повышенный базальный уровень NO производства у MDX мышей, и его освобождение от не - мышечных типов клеток (рис. 2). Поскольку известно, что экспрессия сарколеммой нОАС теряется у MDX мышей [46] , и активность [47] и уровень экспрессии остается неизменным в сердечной и скелетных мышцах MDX мышей или у пациентов с МДД, высокий базальный уровень NO, вероятно, активируется индуцируемой NOS ( иСОА ) [48]. Дальнейшие эксперименты должны подтвердить это понятие. Путем дополнения NO мышечных волокон MDX мышей, приток крови к мышце был усовершенствован и повреждения мышечных волокон индуцированные сокращением были предотвращены (рис. 4). Те же результаты были подтверждены путем применения кленбутерола или 8 - CPT цГМФ. Защита миофибрилл 8 - CPT цГМФ, скорее всего, происходит через сосудистый контроль, так как его преимущества были полностью отменены дополнительным добавлением ATII. Это тормозящее действие ATII, определяется, скорее всего, через сосудистую регуляцию, но не является его прямой катаболической функцией на мышечные волокна, потому ATII сам по себе не вызывает повреждение мышечных волокон. Эти эксперименты предложили главную роль в функциональной ишемии мышечных волокон у MDX мышей. Возможно участие другого вазодилататора, EDHF. Предыдущие исследования EDHF преимущественно осуществляются на сосудистой системе и в меньшей степени на скелетных миоцитах. Там было много мнений, как к личности EDHF, так и его генерации, его действия, его существовании в качестве ( а) секретируемых молекул. Эти исследования приводят к пониманию, что EDHF является гетерогенным субъектом сосудорасширяющим молекул / факторов,
![]()
поведение которых варьируется в зависимости от ткани исследовании. Действие апамина и карибдотоксина, двумя хорошо зарекомендовавшими фармакологическими ингибиторами EDHF, также не было спорным до недавнего времени. Ранее, как считалось, они работали на функции EDHF путем ингибирования гиперполяризации клеток гладких мышц сосудов. В естественных условиях сосудистого эксперимента, однако, все больше доказательств [34] , [35] предполагает, что апамин и карибдотоксин влияют на эндотелиальные клетки, а не на клетки гладких мышц сосудов. Наши данные показывают, что скелетные миоциты могут быть другой точкой действия этих ингибиторов. EDHF, во взаимодействии с NO, может работать, чтобы передать сигналы для мышечно - сосудистой связи, хотя действие обнаруженного Н2О2EDHF должно быть подтверждено. Причина известного производства флуоресцентных сигналов от не - миоцитов (без волокон, как, точечное окрашивание) только в анализах на NO ( 2а, первый и второй столбцы, стрелки ), но не для Н2О2 ( третий и четвертого столбцов ) может быть отчасти потому, что проникают клетки такие, как макрофаги. Количественные доказательства "два хита" механизмов обеспечиваются сравнением двух моделей животных: функциональной модели ишемии у мышей MDX против тяжелой модели ишемии у контрольных мышей. Количественное сравнение контроля и MDX мышах показали, что ингибирование NO / EDHF у одних наблюдалось их функциональная ишемия сравнима с мышами MDX (рис. 6), но не вызывало гибели мышечных клеток в той же степени, как было замечено у MDX мышей. Для стимулирования сокращений для повреждений мышечных волокон у контрольных мышей в сопоставимых масштабах более тяжелой ишемии и более напряженные тетанические стимулы были необходимы (рис. 5 и 6). Эти результаты позволяют предположить, что независимо от ненормального ответа кровотока, мышечные волокна у MDX мышей уже уязвимы к механическим воздействиям. Наши эксперименты показали существование, по крайней мере, двух причин, приводящих к уменьшению повреждений мышечных волокон у мышей MDX: (I) фармакологически поддается лечению фактор ( поток ), который опосредован NO / EDHF и, возможно, другими молекулами, и (II) элементы, зависимые от NO / EDHF или регулирования кровотока ( вспомогательная информация Рисунок S8a - D). Эти два фактора можно объяснить как ранее предложили "два удара " механизма при этом заболевании [23]. Один из этих двух факторов только вызывает повреждение мышц, что наблюдается у MDX мышей. Возможно, млекопитающие развивались так, что есть избыточные факторы, которые предусмотрены для сокращения мышц - индуцированного увеличения потока крови и защиты от сокращения индуцированной стрессом. Таким образом, даже если EDHF синтез / функция завершается ошибкой, еще одним фактором, например, не может заменить в его отсутствие, и наоборот. Пополнение NO, может само по себе, нормализуют кровоток и предотвратить ишемию, несмотря на возможность того, что пораженные мышцы все еще по существу восприимчивы к повреждениям, вызванных сокращениями. Ингибиторы ФДЭ-5 могут быть потенциальным терапевтическим агентом для того чтобы лечить МДД. Дополнительная поддержка, объединяющая данные регуляции сосудов при мышечной дистрофии, продемонстрировала эффективное лечение MDX мышей ингибитором ФДЭ-5 , тадалафилом. Эти данные согласуются с экспериментами в естественных условиях микроскопии, показывающей существенную роль функциональной ишемии в патогенезе мышечной дистрофии, и ожидается, что этот препарат, а также другие вазорегуляторные молекулы могут быть будущей терапевтической мишенью этой болезни. Хотя вазоактивные агенты (NO, 8CPT - цГМФ, и кленбутерол ) почти полностью предотвратили повреждение мышечных волокон в наших краткосрочных экспериментах в естественных условиях от микроскопии (рис. 4), по-прежнему остающееся повреждение мышечных волокон наблюдается у некоторых животных, получавших тадалафил (7а и рис 7б). Это может быть связано с подавлением эффекта препарата после нескольких недель лечения или из-за возможных изменений в объеме движения отдельных животных, которые требуют дальнейших исследований. В этом исследовании мы поставили приоритет биологического вопроса об эффективности тадалафила и начали медикаментозное лечение до рождения и в период лактации и отъема до 4 - недельного возраста, основанное на том, что повреждение мышечного волокна всегда активируется у MDX мышей и пациентов с МДД еще до рождения [53], [54] и, что этот препарат обладает высокой трансплацентарной возможностью распределения [55]. Количество мышечных волокон с центральными ядрами, характерной чертой активной регенерации сократилось после лечения тадалафила. Это наблюдение показывает, что PDE5I может улучшить развитие болезни главным образом через его защитную функцию мышечных волокон, но не через активацию регенерации и, что регенерация мышечных волокон, часто наблюдаемая у мышей MDX, может произойти и вторично активируется дегенерацией мышц. Чтобы полностью установить терапевтический потенциал тадалафила, однако, долгосрочное терапевтическое исследование после отъема мышей MDX не требуется. Наши данные показывают, что защитный эффект цГМФ в острой фазе эксперимента, вероятно, через сосудистую роль, но не исключает возможности того, что NO - цГМФ путь оказывает анаболическое действие через не сосудистые механизмы. Кроме того, наши заключения основной роли сосудистой роли в патогенезе мышечной дистрофии не отрицает или исключает возможности принятые в настоящее время теории « мембраны уязвимости». На самом деле, мембранная уязвимость может быть другим механизмом.

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 |
