
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
![]()
Лечение АЦ снижает ядерную экспрессию NF-kB белка у MDX мышей.
NF-kB обычно находится в цитозоле, но при активации определенных стимулов, в том числе АФК, транслоцируется в ядро ??и связывается с ДНК для регуляции экспрессии генов. Поэтому нам было интересно посмотреть, если АЦ может снизить активацию NF-kB в мышцах MDX. В этих экспериментах мы использовали Вестерн-блоттинг для количественной оценки ядерной транслокации NF-субъединицы белка p65. Как показано на рис. 5A, p65 уровни были значительно больше для мышц MDX контроля, чем для дикого типа, тогда как АЦ значительно снижал экспрессию белка в MDX мышцах. 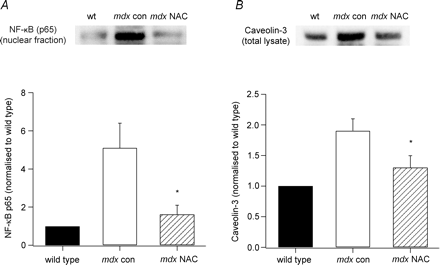
Лечение АЦ снижает кавеолин-3 и увеличивает содержание ?-дистрогликана и атрофина в сарколемме у MDX мышей.
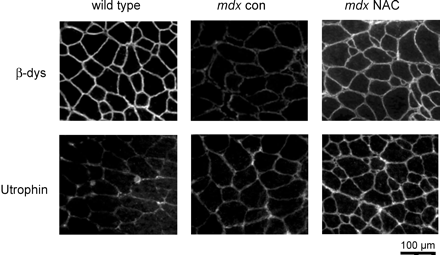
В этих экспериментах, мы хотели определить, какие вещества были вовлечены в регуляцию экспрессии дистрофина-ассоциированных белков. Вестерн-блоттинг показал, что кавеолин-3 у MDX мышей были примерно вдвое больше, чем у дикого типа, и лечение вело к снижению его уровня у мышей MDX (фиг.5В). Когда объединенные данные были нормированы на показатели дикого типа, MDX мышцы контроля были 1,9 ± 0,2 раз больше (N = 6), и это значение было значительно сокращено путем обработки АЦ до 1,3 ± 0,2 (р <0,05, N = 5). EDL сечение от дикого типа, MDX контроля и MDX АЦ-обработанных мышей подвергали иммунному окрашиванию с антителами к кавеолину-3, ?-дистрогликану и атрофину. Что подтвердило результаты вестерн-блоттинга, мышечные секции АЦ-обработанных мышей MDX показали менее интенсивное окрашивание к кавеолину-3 по сравнению с MDX контроля (см. рис. 3А). Мы также обнаружили, что сарколемное окрашивание ?-дистрогликана и атрофина было увеличено для леченных мышц по сравнению с контрольными мышами MDX (рис. 6). Как и ожидалось, окрашивания атрофина было незначительным у дикого типа мышц.
![]()
Обсуждение.
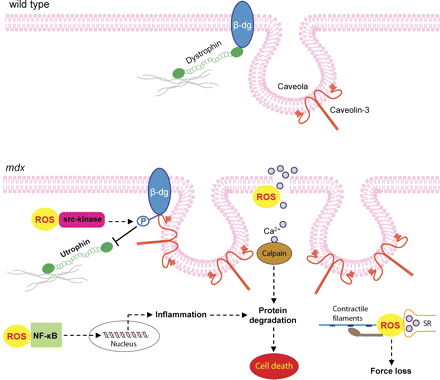
Существует немало свидетельств окислительного повреждения дистрофических мышц, а также положительное влияние антиоксидантов (Мессина и др. 2006;. Hnia и др. 2007 год.). Дистрофин-дефицитные клетки более чувствительны к АФК-индуцированному повреждению (Rando соавт. 1998) и есть свидетельства регуляции эндогенных антиоксидантных систем обороны, приводящей к повышенной продукции АФК (Tidball & Велинг-Henricks, 2007). В текущем исследовании, мы использовали антиоксидант АЦ для получения новых данных, которые увеличили понимание роли АФК в остром повреждении мышц, вызванным сокращениями при растяжении, и в рамках текущего дегенеративного процесса, который происходит естественно в мышцах интактных мышей MDX. Мы также обнаружили, что АФК играют важную роль в регуляции дистрофин-ассоциированных белков, которая может быть отменена путем обработки АЦ. Наша интерпретация этих результатов представлена на рис. 7. Данные, представленные в этой статье соответствуют недавним результатам нашей лаборатории, в которой АЦ применялся для улучшения функции левого желудочка и мышцы, уменьшал признаки поражения сердца и дегенерации у мышей MDX (Williams & Allen, 2007).
АЦ предотвращает стресс-индуцированную проницаемость мембран в мышцах MDX
Это широко распространенное мнение, что поверхностная мембрана дистрофических мышц более хрупкая, чем у нормальных мышц, что тем самым делает ее более восприимчивой к микро повреждениям во время мышечного сокращения, особенно при сокращениях после растяжений (Moens и др. 1993;. Petrof соавт. 1993). Тем не менее, мы недавно сообщили, что большую часть повышенной проницаемости мембраны после сокращений мышц MDX при комнатной температуре можно предотвратить путем применения стрептомицин SAC блокаторов и GsMTx4 (Whitehead и соавт. 2006a). Учитывая, что эти препараты также предотвращают стресс-индуцированный
![]()
рост внутриклеточного Ca 2 + в MDX волокнах (Енг и др.. 2005), мы предположили, что пути Ca 2 +-зависимых повреждений были наиболее вероятной причиной повышенной проницаемости мембраны. В текущем исследовании, результаты экспериментов на изолированной мышце показывают, что АЦ препятствует повышенной проницаемости мембран, связанной с сокращениями в растянутых мышцах MDX при 35 ° С. Таким образом, это дает дополнительные свидетельства, показывающие, что индуцированное увеличение проницаемости мембраны в мышцах MDX не является первичным следствием механической травмы, а скорее возникает из-за эффектов АФК и / или Ca2 + притока через УКД (Whitehead и соавт. 2006a). Хотя число СКЭ положительных волокон в данном исследовании является относительно небольшим по сравнению с большим дефицитом силы, они являются надежными показателями повреждения. СКЭ поглощение отражает значительное увеличение проницаемости мембраны от небольшой популяции серьезно поврежденных, пренекротических волокон (Хамер и др., 2002;.. Уайтхед и др. 2006a), тогда как большинство волокон может исключить СКЭ и остаются возбудимыми, но все еще повреждены и имеют большой дефицит силы (Енг и др.. 2005).
АФК вклад в стресс-индуцированную силу сокращения мышц у MDX.
Отличительной особенностью повреждений, связанных с растянутыми нормальносокращающимися мышцами, является сохранение снижения силы в течение нескольких дней (Morgan & Allen, 1999). Как и ожидалось, снижение силы было значительно больше для MDX мышц по сравнению с диким типом (Moens и др. 1993;. Уайтхед и др. 2006a.). Важно отметить, что АЦ обеспечивал значительную защиту силы сокращения мышц у MDX, но не у дикого типа. Это говорит о том, что чрезмерная продукця АФК и окислительное повреждение при сокращениях не является неотъемлемой чертой нормальных скелетных мышц, а скорее аномалия, связанная с дистрофическими мышцами. Пока неясно, как увеличение АФК вызвало снижение мышечной силы в наших экспериментах, но хорошо известно, что АФК могут окислять сократительные белки и белки возбуждения-сокращения (ЕС) в скелетных мышцах, что окислительно-восстановительный баланс влияет на способность мышц вырабатывать силу (Reid, 2001 ). Кроме того, увеличение АФК опосредованной проницаемости мембран может также вносить вклад в потерю силы у MDX мышц через приток Na +, который может вызвать деполяризацию мембраны и уменьшить возбудимость мышечных волокон.
Лечение АЦ уменьшает повреждения мышц MDX в естественных условиях.
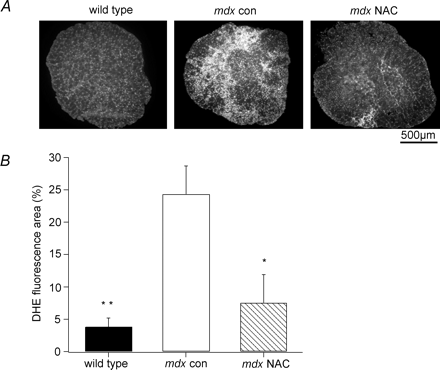
После 6 недель орального приема АЦ, мышцы у мышей MDX показали заметное снижение повреждений. Наличие центральных ядер в мышечных волокнах является хорошо установленным показателем регенерации после повреждения мышцы (McGeachie соавт. 1993). Лечение АЦ значительно сократил количество мышечных волокон MDX с центральными ядрами, подразумевая, что прогрессирование повреждения мышц было замедлено. Уровни АФК существенно возросли в MDX мышцах и значительно ослаблялись лечением (см. рис. 4). Это говорит о том, что повышение окислительного повреждения является важным фактором в течение этого длительного периода (3-9 недельного возраста) мышечной дистрофии у MDX мышей. ДГЕ окрашенные срезы мышц мышей MDX также показали области плотно упакованых, интенсивно окрашенных клеток, которые, возможно, являются воспалительными клетками, такими как макрофаги и нейтрофилы. Эти клетки, как известно, производят существенные концентрации АФК при участии НАДФ оксидазы, которые могут вызвать лизис мышечных клеток в воспалительных каскадах (Nguyen & Tidball, 2003). Таким образом, воспалительные клетки могут обеспечить дополнительный источник АФК в мышцах MDX, который усугубляет уже существующие повреждения, вызванные внутриклеточными АФК-опосредованными путями.
АЦ лечение ингибирует экспрессию NF-kB у MDX мышей.
NF-kB активации обусловлено различных клеточных стимулов, в том числе АФК. При активации NF-kB транслоцируется в ядро ??и регулирует транскрипцию многих генов (Kumar и соавт. 2004). NF-kB связывания ДНК было показано, что возросла во много раз в мышцах от пациентов с МДД (Monici соавт. 2003) и MDX мышей (Мессина и соавт. 2006), даже в молодом возрасте (15 дней), задолго до начала измеримых повреждения мышц (Kumar & Boriek, 2003). Это согласуется с увеличением окислительного стресса в мышцах молодых MDX (Disatnik соавт. 1998) и, следовательно, АФК-индуцированная активация NF-kB, вероятно, будет фактором, способствующим обширной волне повреждения мышц, которая следует этому период покоя. Среди генов, регулируемых NF-kB, ряд, как было показано, участвуют в дистрофическом процессе заболевания, включая провоспалительные цитокины, таких как ФНО-(Основание и Torrisi, 2004;. Hodgetts и др., 2006), и матриксные металлопротеиназы, которые вызывают расщепления ?-дистрогликана (Hnia соавт. 2007). Наши результаты показывают, что лечение АЦ значительно снижает экспрессию ядерного NF-p65 белка, что показывает, что АФК являются важным медиатором NF-kB активации и транслокации в ядро. Наши результаты также согласуются с недавними работами, которые показывают, что NF-kB ДНК связывающая активность значительно снижается у MDX мышей, получавших лечение антиоксидантом (Мессина и др. 2006;.. Hnia и др. 2007, Фарид и др. 2005 ).
Лечение АЦ регулирует экспрессию дистрофин-ассоциированных белков, в то время как большинство белков комплекса регулируются по принципу отрицательной связи кавеолином-3, что представляет собой заметное исключение, с уровнем приблизительно в 2 - 3 раза выше у MDX (Vaghy и др. 1998 год, Repetto и соавт. 1999). В наших экспериментах вестерн-блоттинг показал, что экспрессия кавеолина-3 снизилась при лечении АЦ у MDX мышей, и это сопровождалось повышенной экспрессией ?-дистрогликана и гомолога дистрофина, атрофина на сарколемме (рис. 6). Это говорит о том, что АФК-опосредованные пути способствуют нарушению регуляции этих белков в MDX мышцах. Мы предполагаем, что кавеолин-3 может играть важную роль в регулировании этой реконструкции комплекса ДАГ в дистрофических мышцах. Трансгенная сверхэкспрессия кавеолина-3 у мышей приводит к понижающей регуляции ?-дистрогликана и дистрофина, и производит МДД-подобные мышечные

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 |
