
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
ОБСУЖДЕНИЕ
Хотя в настоящее время нет эффективных терапевтических возможностей для пациентов с МДД, есть много перспективных активных областей исследований ( 37,38 ). Большинство из этих вариантов сосредоточены на восстановлении механической стабильности мышцы путем клеточной терапии, генной терапии или сочетания того и другого. Недавно было показано, что смягчение проблем, связанных с сосудистым снабжением у MDX мышей может существенно улучшить дистрофический фенотип ( 5,12,19 ) . Однако взаимосвязь развития между ангиогенезом и мышечным фенотипом дистрофии предстоит выяснить. В этом
![]()
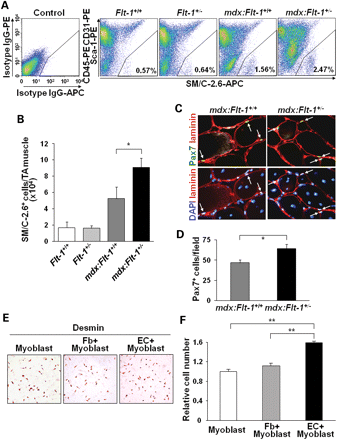
докладе мы стремились компенсировать функциональную ишемию в мышцах мышей с МДД при помощи увеличения плотности уровня развития сосудов. Мы обнаружили, что Flt - 1 + / - мыши, а также MDX: Flt - 1 + / - мыши показывают повышенную плотность кровеносных сосудов в мышцах по сравнению с контролем Flt - 1 + / + или MDX: Flt - 1 + / + мышами. Увеличение плотности кровеносных сосудов приводит к увеличению перфузии крови в мышцах и улучшения мышечного гистологического фенотипа, как следствие улучшение сократительной функции мышц. Одновременно это также привело к увеличению выживаемости и улучшению мышечной гистологии у MDX: utrn - / - : Fit - 1 + / - в сочетании с тройными мутантными мышами по сравнению с контрольными MDX: utrn - / - : FLT - 1 + / + двойными мутантными мышами. Насколько нам известно, это исследование является первым, показывающим, что с развитием увеличения ангиогенеза и тем самым, увеличения сосудистой плотности, может частично уменьшится дистрофический фенотип у мышей с МДД. Потенциальные механизмы включают (I) снижение воспалительной реакции, (II) автономные эффекты клеток, (III) уменьшение ишемии путем увеличения перфузии ткани или (IV) расширение взаимодействия сосудистой сети для увеличения пролиферации спутниковых клеток и / или защиты от повреждения мышц. Сообщалось также, что, чтобы освободить место для новых мышечных волокон, повреждение мышц сопровождается приходом макрофагов, чтобы убрать продукты распада клеток, оставленные поврежденной мышечной тканью ( 39-42 ) . Тем не менее, мы не нашли никакого существенного различия в численности Mac-1 + - или GR-1 + - клеток проникающих в мышцу MDX: Fit - 1 + / - по сравнению с контрольной MDX: Fit - 1 + / + мышей. Так как наша система модулирует экспрессию FLT - 1 , важно исследовать воздействие на автономные мышечные волокна и миогенные клетки-предшественники. Во-первых, срезы мышц показали, что Flt - 1 - или Flk -1- экспрессирующие клетки расположены за пределами базальной пластинки мышечных волокон. Мы также решили исследовать экспрессию и спутниковым клеток, и полученных миобластов, и культур мышечных волокон, так как возможно получить чистые популяции спутниковых клеток с помощью этого метода. При иммунологическом обнаружении LacZ у мышей, наши данные ясно показывают, что мышечные волокна, спутниковые клетки и полученные миобласты не экспрессируют FLT-1 и FLK - 1. Это означает, что нет никаких автономных клеточных эффектов на мышечные волокна или миогенные клетки-предшественники посредством этих рецепторов в нашей системе. Важно отметить, что увеличение плотности кровеносных сосудов у Fit - 1 + / - и MDX : Fit - 1 + / - мышей также переведены на увеличение LDF в состоянии покоя. Во время отдыха скелетным мышцам не хватает полной перфузии из-за отсутствия полного набора микрососудов (44). Это набор микрососудов был рассмотрен во время физических упражнений, а также во время инфузии инсулина. Во время мышечной деятельности NOS - опосредованная вазодилатация может увеличить градиент пассивного давления в капиллярах, таким образом, увеличивая работу микроциркуляторного русла и повышая тканевую перфузию ( 45,46 ). Ишемия, созданная в связи с отсутствием такого расширения сосудов при сокращении, является прямой причиной повреждения мышечных волокон в естественных условиях у MDX мышей (12). Таким образом, увеличение базального кровотока обнаруженного в наших моделях мышечной дистрофии может предложить защитный механизм для сокращения повреждения (43). Увеличение производства эндотелиальных клеток в мышцах может иметь множество различных благотворных эффектов на дистрофические мышцы через паракринную стимуляцию, защищая поврежденные мышечные волокона и активируя миогенные клетки-предшественники. Многочисленные отчеты показали, перекрестные помехи между эндотелиальными клетками и клетками-сателлитами (19,47,48 ). Наши результаты, а также другие показали, что эндотелиальные клетки и факторы, производимые в них могут выступить посредником ответа на выживание спутниковых клеток и пролиферацию(18,47,48). Это в соответствии с нашими экспериментами с культурами, которые показывают увеличение пролиферации клеток, полученных спутниковых миобластов, когда их совместно культивировали с эндотелиальными клетками. Недавно Христов и др. . (47) показали, что клетки-сателлиты преимущественно расположены рядом с капиллярами. Так как наша модель увеличивает количество капилляров, находящихся в мышцах, что может эффективно увеличить количество сосудистых ниш. В этом докладе мы показываем, что число сателлитных клеток и миогенных клеток-предшественников заметно увеличивается у MDX: Fit - 1 + / - мышей по сравнению с контрольными MDX: Fit - 1 + / + мышами. Это увеличение числа спутниковых клеток и миогенных клеток-предшественников могут быть ответственны за фенотип MDX: Flt - 1 + / - мышей. Данное исследование показывает, что увеличение ангиогенеза может быть новым подходом для улучшения некоторых дистрофинопатий и функциональных параметров, связанных с МДД, которые можно использовать в конъюнктуре с другими стратегиями. Например, внутримышечная инъекция VEGF, содержащие рекомбинантные аденоассоциированные вирусные векторы, как было показано, чтобы функциональные и гистологическое улучшение в ишемических мышцах и мышцах MDX (19,49,50). Это улучшение видно в сочетании с увеличением ангиогенеза в мышце. Ито и др. (5) также показали важность отсутствия дистрофина в гладких мышцах сосудов. Они показали, что экспрессия дистрофина в гладкомышечных клетках MDX мышей привело к частичному улучшению фенотипа мышц. Важно понять, как васкуляризация взаимодействовует с мышечными волокнами и мышечными стволовыми клетками, чтобы обеспечить необходимый кислород и питание, защищать от гибели клетки и обеспечивают сосудистую нишу. Необходимы дальнейшие исследования, чтобы установить точный механизм и
![]()
долгосрочные последствия увеличения васкуляризации. С терапевтической точки зрения, неоваскуляризация может быть индуцирована в дистрофических мышцах с использованием введения ангиогенных факторов, таких как VEGF белок.
Ссылки
↵ Dalkilic I., Kunkel L. M. Muscular dystrophies: genes to pathogenesis. Curr. Opin. Genet. Dev. 2003;13:231-238. doi:10.1016/S0959-437X(03)00048-0. CrossRef Medline Web of Science ↵ Davies K. E., Nowak K. J. Molecular mechanisms of muscular dystrophies: old and new players. Nat. Rev. Mol. Cell Biol. 2006;7:762-773. doi:10.1038/nrm2024. CrossRef Medline Web of Science ↵ Mendell J. R., Engel W. K., Derrer E. C. Duchenne muscular dystrophy: functional ischemia reproduces its characteristic lesions. Science 1971;172:1143-1145. doi:10.1126/science.172.3988.1143. Abstract/FREE Full Text ↵ Miyatake M., Miike T., Zhao J., Yoshioka K., Uchino M., Usuku G. Possible systemic smooth muscle layer dysfunction due to a deficiency of dystrophin in duchenne muscular dystrophy. J. Neurol. Sci. 1989;93:11-17. doi:10.1016/0022-510X(89)90157-3. CrossRef Medline Web of Science ↵ Ito K., Kimura S., Ozasa S., Matsukura M., Ikezawa M., Yoshioka K., Ueno H., Suzuki M., Araki K., Yamamura K., et al. Smooth muscle-specific dystrophin expression improves aberrant vasoregulation in mdx mice. Hum. Mol. Genet. 2006;15:2266-2275. doi:10.1093/hmg/ddl151. Abstract/FREE Full Text ↵ Cullen M. J., Jaros E. Ultrastructure of the skeletal muscle in the X chromosome-linked dystrophic (mdx) mouse: comparison with duchenne muscular dystrophy. Acta Neuropathol. 1988;77:69-81. doi:10.1007/BF00688245. CrossRef Medline ↵ Loufrani L., Levy B. I., Henrion D. Defect in microvascular adaptation to chronic changes in blood flow in mice lacking the gene encoding for dystrophin. Circ. Res. 2002;91:1183-1189. doi:10.1161/01.RES.0000047505.11002.81. Abstract/FREE Full Text ↵ Loufrani L., Matrougui K., Gorny D., Duriez M., Blanc I., Levy B. I., Henrion D. Flow (shear stress)-induced endothelium-dependent dilation is altered in mice lacking the gene encoding for dystrophin. Circulation 2001;103:864-870. Abstract/FREE Full Text ↵ Weir A. P., Burton E. A., Harrod G., Davies K. E. A - and B-utrophin have different expression patterns and are differentially up-regulated in mdx muscle. J. Biol. Chem. 2002;277:45285-45290. doi:10.1074/jbc. M205177200. Abstract/FREE Full Text ↵ Coral-Vazquez R., Cohn R. D., Moore S. A., Hill J. A., Weiss R. M., Davisson R. L., Straub V., Barresi R., Bansal D., Hrstka R. F., et al. Disruption of the sarcoglycan–sarcospan complex in vascular smooth muscle: a novel mechanism for cardiomyopathy and muscular dystrophy. Cell 1999;98:465-474. doi:10.1016/S0092-8674(00)81975-3. CrossRef Medline Web of Science ↵ Rando T. A. Role of nitric oxide in the pathogenesis of muscular dystrophies: a ‘two hit’ hypothesis of the cause of muscle necrosis. Microsc. Res. Tech. 2001;55:223-235. doi:10.1002/jemt.1172. CrossRef Medline Web of Science ↵ Asai A., Sahani N., Kaneki M., Ouchi Y., Martyn J. A., Yasuhara S. E. Primary role of functional ischemia, quantitative evidence for the two-hit mechanism, and phosphodiesterase-5 inhibitor therapy in mouse muscular dystrophy. PLoS ONE 2007;2:e806. doi:10.1371/journal. pone.0000806. CrossRef Medline ↵ Fong G. H., Rossant J., Gertsenstein M., Breitman M. L. Role of the flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature 1995;376:66-70. doi:10.1038/376066a0. CrossRef Medline ↵ Fong G. H., Zhang L., Bryce D. M., Peng J. Increased hemangioblast commitment, not vascular disorganization, is the primary defect in flt-1 knock-out mice. Development 1999;126:3015-3025. Abstract ↵ Ferrara N., Gerber H. P., LeCouter J. The biology of VEGF and its receptors. Nat. Med. 2003;9:669-676. doi:10.1038/nm0603-669. CrossRef Medline Web of Science ↵ Kearney J. B., Ambler C. A., Monaco K. A., Johnson N., Rapoport R. G., Bautch V. L. Vascular endothelial growth factor receptor flt-1 negatively regulates developmental blood vessel formation by modulating endothelial cell division. Blood 2002;99:2397-2407. doi:10.1182/blood. V99.7.2397. Abstract/FREE Full Text ↵ Springer M. L., Ozawa C. R., Banfi A., Kraft P. E., Ip T. K., Brazelton T. R., Blau H. M. Localized arteriole formation directly adjacent to the site of VEGF-induced angiogenesis in muscle. Mol. Ther. 2003;7:441-449. doi:10.1016/S1525-0016(03)00010-8. CrossRef Medline Web of Science ↵ Arsic N., Zacchigna S., Zentilin L., Ramirez-Correa G., Pattarini L., Salvi A., Sinagra G., Giacca M. Vascular endothelial growth factor stimulates skeletal muscle regeneration in vivo. Mol. Ther. 2004;10:844-854. doi:10.1016/j. ymthe.2004.08.007. CrossRef Medline Web of Science ↵ Messina S., Mazzeo A., Bitto A., Aguennouz M., Migliorato A., De Pasquale M. G., Minutoli L., Altavilla D., Zentilin L., Giacca M., et al. VEGF overexpression via adeno-associated virus gene transfer promotes skeletal muscle regeneration and enhances muscle function in mdx mice. FASEB J. 2007;21:3737-3746.![]()

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 |
