

Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
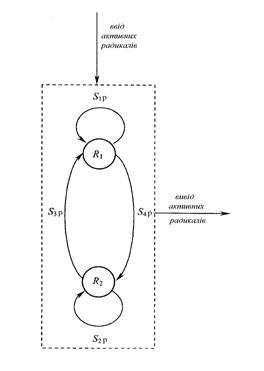
u1 = S1p, Δ1 = kp11 [A1], M(1) = M1 = kp12 [A1],
u2 = S2p, Δ2 = kp22 [A2], M(2) = M2 = kp21 [A2], (6)
u3 = S3p + S4p, Δ3= kp12 kp21 [A1][A2], M(3) = M(1,2) = 1.
| Вирази для швидкостей за маршрутами мають вигляд: Враховуючи стехіометрію маршрутів, швидкості кополімеризації за кожним компонентом дорівнюють d[A1] / d t = r1 + r2, d[A2] / d t = r1 + r3 . (8) Таким чином, рівняння швидкості кополімеризації за і–тим компонентом при лінійному обриванні ланцюга можна записати у вигляді Тут, однак, концентрації мономерів [Asi] різні за об’ємом мікрозерен ПМФ і залежать не тільки від часу виділення даної порції ПМФ, швидкості кополімеризації в ній, а й від швидкості надходження даного мономера із МПФ у ПМФ. |
Рис. 3. Граф G = {R, S}, що відображає топологію кінетичної схеми розвитку ланцюга при кополімеризації |
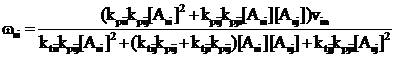 . (9)
. (9)5. Розглядаючи ситуацію в момент часу τ ≥ t*, в інтервалі τ + dτ з МПФ виділиться елементарний об’єм ПМФ, рівний vodφs. Приймаємо, що він однорідний за складом і питома швидкість кополімеризації в ньому підкоряється рівн. (9).
Із рівняння балансу по обох мономерах можна одержати
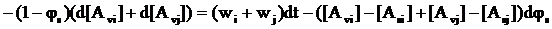 , (10)
, (10)
де через Wi позначена швидкість кополімеризації в МПФ і міжфазному шарі
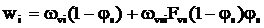 . (11)
. (11)
Із цих виразів випливає:
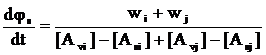 . (12)
. (12)
Тут [Avi] і [Asi], відповідно, концентрації і–го мономера в МПФ і ПМФ в момент часу виділення даної порції dφs полімер–мономерної фази.
6. У двофазній полімеризаційній системі повний опис кінетики процесу вимагає знання швидкостей витрати мономерів у кожній фазі.
Вираз для швидкості зміни концентрації і–го мономера в МПФ і міжфазному шарі знайдено із рівн. (10):
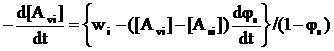 . (13)
. (13)
Швидкість зміни середньої ![]() за об’ємом мікрозерен концентрації і–го мономера в ПМФ описано виразом
за об’ємом мікрозерен концентрації і–го мономера в ПМФ описано виразом
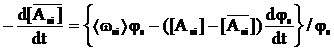 , (14)
, (14)
де 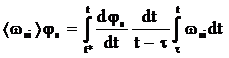 , t* ≤ τ . (15)
, t* ≤ τ . (15)
Показано, що рівняння (12), (13) і (14) складають повну систему диференційних рівнянь, що описують кінетику кополімеризації у трьох реакційних зонах двофазної полімеризаційної системи.
У четвертому розділі представлені результати дослідження стаціонарної кінетики фотокополімеризації біфункційних (мет)акрилатів, а також запропонована кінетична модель стаціонарної тривимірної фотоініційованої кополімеризації ди(мет)акрилатів до глибоких конверсій.
Кінетика стаціонарної фотоініційованої кополімеризації біфункційних (мет)акрилатів до глибоких конверсій вивчена на прикладі системи 1,6–гександіолдиакрилату (ГДДА) та триетиленглікольдиметакрилату (ТГМ–3) при мольних співвідношеннях компонентів 4:1, 2:1, 1:1, 1:2 і 1:4 в залежності від концентрації фотоініціатора (1 і 2 % мол.) та інтенсивності УФ–опромінення (7, 17 і 48 Вт/м2).
Частина експериментальних кінетичних кривих кополімеризації ди(мет)акрилатів ГДДА і ТГМ–3 в залежності від складу системи показана на рис. 4.
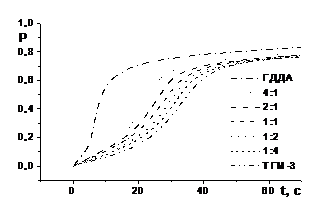
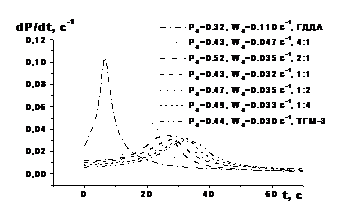
Рис. 4. Інтегральні кінетичні криві і їх диференціальні анаморфози фотоініційованої кополімеризації системи ГДДА – ТГМ–3 залежно від її складу: [IR 651] = 2,0 % мол., Е0 = 7 Вт/м2, Т = 293 К
Як видно, кінетичні криві фотоініційованої кополімеризації 1,6–гександіолдиакрилату і триетиленглікольдиметакрилату характеризуються типовим S–подібним виглядом і складаються всього із двох ділянок: автоприскорення і автогальмування. При зміні відношення ГДДА : ТГМ–3 на користь диметакрилату ТГМ–3 кінетичні криві змінюються за типом від характерного для диакрилату ГДДА до характерного для диметакрилату ТГМ–3. Головна роль в зміні типу кінетичних кривих належить диметакрилату ТГМ–3.
Крім того, з ростом долі ТГМ–3 у вихідній композиції різко збільшується конверсія Ро, якій відповідає максимальна швидкість Wo кополімеризації, при одночасному зниженні Wo. Це свідчить про зниження вкладу міжфазного процесу в сумарний, що може бути викликано двома причинами. Перша полягає в тому, що низька константа швидкості kpii росту ланцюга гомополімеризації ТГМ–3 приводить і до низьких значень перехресних констант kpij. Друга причина пояснюється різною структурою міжфазного шару на границі мономер–полімерного розчину з мікрозернами ТГМ–3 і ГДДА.
Кінетична модель тривимірної фотоініційованої кополімеризації біфункційних (мет)акрилатів до глибоких конверсій
У сітчастому полімері розчинність мономера мала, тому процесом полімеризації в полімерній фазі можна знехтувати. Отже, процес полімеризації поліфункційних мономерів реалізується в двох реакційних зонах: об’ємі рідкої мономерної фази (МФ), в якій практично відсутній розчинений полімер, і міжфазному шарі на границі МФ з твердою полімерною фазою (ПФ), в якій мала розчинність мономера.
Тому загальну швидкість витрати і–го мономера в розрахунку на весь об’єм системи можна записати у вигляді
![]() . (16)
. (16)
Тут ωvi і ωvsi – питомі парціальні швидкості кополімеризації і–го мономера в об’ємах МФ і ПФ відповідно; φs – біжуча об’ємна доля мікрозерен ПФ.
В об’ємі рідкої мономерної фази кополімеризація проходить за класичною кінетичною схемою з квадратичним обриванням ланцюга (2). Тому питомі швидкості кополімеризації в МФ описуються тим самим рівнянням Майо–Уоллінга (3).
Тверда ПФ на границі з рідкою МФ утворює жорстку структуру міжфазного шару. Тому в міжфазному шарі на границі МФ і ПФ сегментальна і трансмісійна рухомість макрорадикалів різко зменшується, збільшуючи роль дифузійного гальмування квадратичного обривання ланцюга. В той же час збільшується ймовірність захоплення або самозахоронення зростаючого макрорадикала в полімерній матриці. Тому приймаємо, що в міжфазному шарі обривання ланцюга є лінійним і проявляє себе як акт росту макрорадикала, який веде в пастку, що відображено кінетичною схемою (5). В цьому випадку питомі швидкості ωvsi кополімеризації за і–тим мономером в об’ємі реакційної зони міжфазного шару можна описати наведеним вище рівнянням (9).
Вираз для швидкості dφs / dt виділення твердої ПФ одержано із виведеного раніше рівняння (12), припустивши, що в ньому концентрації мономерів в ПФ близькі до нуля: [Asi] ≈ 0, [Asj] ≈ 0:
dφs / dt = (wi + wj) / ([Avi] + [Avj]). (17)
В сукупності рівняння (3), (9), (16) і (17) повністю описують кінетику кополімеризації двох біфункційних мономерів до глибоких конверсій в моделі двох реакційних зон.
У п’ятому розділі представлені результати дослідження стаціонарної кінетики фотокополімеризації моно– та біфункційних (мет)акрилатів.
Стаціонарна кінетика фотоініційованої кополімеризації моно– та біфункційних (мет)акрилатів до глибоких конверсій вивчена на прикладі системи триетиленглікольдиметакрилат – гліцидилметакрилат при мольних співвідношенні компонентів ТГМ–3 : ГМА 4:1, 2:1, 1:1, 1:2, 1:4 в залежності від концентрації фотоініціатора (1 і 2 % мол.) та інтенсивності УФ–опромінення (7, 17 і 48 Вт/м2).
Частина експериментальних кінетичних кривих кополімеризації ТГМ–3 і ГМА в залежності від складу вихідної системи показана на рис. 5.
Кінетика кополімеризації моно– і біфункційного мономерів композиції ТГМ–3 : ГМА виявляє в залежності від її складу риси, властиві лінійній і тривимірній полімеризації. Як видно із рис. 5, при зміні відношення ТГМ–3 : ГМА на користь диметакрилату ТГМ–3 кінетичні криві змінюються за типом від характерного для монофункційного мономеру ГМА до характерного для біфункційного мономеру ТГМ–3. При цьому різко зменшується конверсія Р0, якій відповідає максимальна швидкість процесу W0, при одночасному зростанні W0.
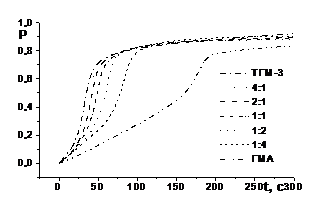

Рис. 5. Інтегральні кінетичні криві і їх диференціальні анаморфози фотоініційованої кополімеризації системи ТГМ–3 – ГMА залежно від її складу: [IR 651] = 2,0 % мол., Е0 = 7 Вт/м2, Т = 293 К
Головна роль в зміні типу кінетичних кривих належить диметакрилату ТГМ–3: вже при молярному співвідношенні ТГМ–3 : ГМА = 1 : 2 кінетична крива кополімеризації практично повністю набуває ознак, характерних для полімеризації диметакрилату, тобто, практично не має початкової лінійної ділянки і складається тільки із стадій автоприскорення і автогальмування. З точки зору концепції реакційних зон це означає, що наявність біфункційного комономеру різко зменшує як розчинність кополімеру у вихідній комономерній суміші, так і мономерів у кополімері. Як наслідок, стадія автоприскорення починається при більш низькій конверсії, а внесок в сумарну кінетику процесу кополімеризації в полімер–мономерній фазі зменшується. В той же час при співвідношенні ТГМ–3 : ГМА = 1 : 4, хоч і у цьому випадку утворюється тривимірна сітка, але з довгими ланцюгами між вузлами, має місце порівняно висока розчинність кополімера в вихідній суміші, якій відповідає загальна конверсія Р ≈ 0.3, так що кінетична крива кополімеризації схожа за формою до кінетичної кривої полімеризації ГМА.
У шостому розділі представлені результати дослідження кінетики загибелі метакрилатних та акрилатних макрорадикалів в одержаних полімерних матрицях різної природи, що дозволило одержати числові значення коефіцієнтів дифузії макрорадикалів в полімерних матрицях і оцінити характеристичні часи сегментального руху.
Кінетика загибелі метакрилатних макрорадикалів у полімерних матрицях
ТГМ–3 ТГМ–3 : ГМА = 1:1 ГМА | диметакрилату ТГМ–3, монометакрилату ГМА і їх кополімера ТГМ-3 : ГМА = 1 : 1 досліджена ЕПР–спектроскопічним методом в температурному діапазоні 20 – 70 0С. Типові ЕПР–спектри парамагнітних центрів наводяться на рис. 6. |
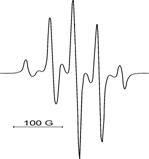

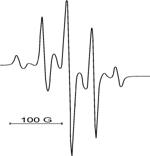
Рис. 6. ЕПР–спектри метакрилових радикалів у різних полімерних матрицях
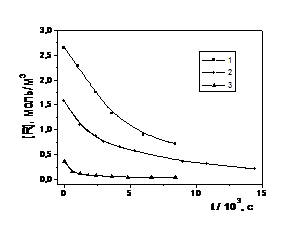
Вихідні кінетичні криві загибелі радикалів, наведені на рис. 7, є типовими для всіх температур, хоч їхні характеристичні часи при зміні температури від 20 до 70 0С зменшуються приблизно на 4 порядки.
|
|
Рис. 7. Вихідні кінетичні криві загибелі радикалів при 60 0С в полімерних матрицях: 1 – ТГМ–3; 2 – ТГМ–3 : ГМА = 1:1; 3 – ГМА | Рис. 8. Представлення кінетичних кривих загибелі радикалів у різних полімерних матрицях в координатах рівняння реакції другого порядку: 1 – ТГМ–3; 2 – ТГМ–3 : ГМА = 1 : 1; 3 – ГМА |
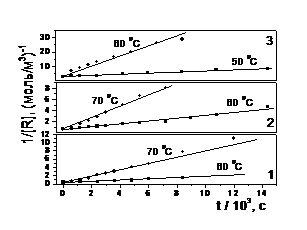
Представлення експериментальних даних в координатах реакції другого порядку (див. рис. 8.)
1 / [R] = 1 / [R]o + kt t, (18)
де [R]о і [R] – початкова і біжуча в часі t концентрації радикалів, свідчить про бімолекулярний механізм їхньої загибелі, не ускладнений залежністю константи швидкості kt від часу. Результати розрахунку kt за рівнянням (18) для всіх трьох досліджуваних полімерних матриць при різних температурах представлені в табл. 1.
Таблиця 1
Експериментальні (kt) і розрахункові (D, τ) значення параметрів загибелі макрорадикалів і їхнього дифузійного перенесення
Система | T, K | kt, м3 /моль·с | D, м2/с | τ, c |
ТГМ–3 | 293 | 3.9×10–7 | 6.9×10–23 | 0.5 |
313 | 1.7×10–5 | 3.0×10–21 | 1.3×10–2 | |
323 | 5.6×10–5 | 1.0×10–20 | 3.8×10–3 | |
333 | 1.1×10–4 | 2.0×10–20 | 2.0×10–3 | |
343 | 8.3×10–4 | 1.5×10–19 | 2.6×10–4 | |
ТГМ–3 : ГМА = 1 : 1 | 293 | 4.0×10–7 | 7.0×10–23 | 0.2 |
323 | 4.8×10–5 | 8.5×10–21 | 1.7×10–3 | |
333 | 2.5×10–4 | 4.4×10–20 | 3.3×10–4 | |
343 | 1.0×10–3 | 1.8×10–19 | 0.8×10–4 | |
ГМА | 293 | 2.7×10–6 | 4.8×10–22 | 1.5×10–2 |
313 | 6.2×10–5 | 1.1×10–20 | 6.8×10–4 | |
323 | 3.9×10–4 | 6.9×10–20 | 1.1×10–5 | |
333 | 3.5×10–3 | 6.2×10–19 | 1.2×10–5 |
За температурною залежністю kt (див. рис. 9.) визначені числові значення параметрів рівняння Арреніуса

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 |
