
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Министерство здравоохранения РФ
Уральская государственная медицинская академия дополнительного образования
Кафедра терапии, клинической фармакологии и эндокринологии
Медицинский центр по коррекции метаболических нарушений и укреплению здоровья
, , ,
Диагностика и лечение дислипидемии у больных
метаболическим синдромом
Учебное пособие
Челябинск 2003
Авторы:
· – доктор медицинских наук, профессор, заведующий кафедрой терапии, клинической фармакологии и эндокринологии УГМАДО;
· – кандидат медицинских наук, ю доцент кафедры терапии, клинической фармакологии и эндокринологии УГМАДО;
· – ассистент кафедры терапии, клинической фармакологии и эндокринологии УГМАДО;
· - ассистент кафедры терапии, клинической фармакологии и эндокринологии УГМАДО.
Рецензент:
· – доктор медицинских наук, профессор, заведующий кафедрой пропедевтики внутренних болезней ЧГМА.
Методические рекомендации предназначены для врачей-терапевтов, кардиологов, клинических фармакологов, эндокринологов, диетологов, врачей общей практики, студентов старших курсов медицинских ВУЗов.
Учебное пособие издано
при поддержке фармацевтической компании KRKA, d. d., Novo mesto Slovenia
Уральская государственная медицинская академия
дополнительного образования
Рассмотрено на заседании УМК. Утверждено
Протокол № 5 от «___» апреля 2003 года на заседании Ученого Совета
24 апреля 2003 года.
Диагностика и лечение дислипидемии у больных
метаболическим синдромом.
Учебное пособие
Челябинск 2003
Содержание.
Классы липопротеинов. Классификация дислипидемий (первичные, вторичные). Диагностика и концепция развития метаболического синдрома. Влияние инсулинорезистентности на метаболизм липидов. Особенности дислипидемии у больных сахарным диабетом. Концепция факторов риска развития сердечно-сосудистых заболеваний. Целевые уровни липидов. Модификация образа жизни. Физическая активность. Лечебное питание при дислипидемиях. Статины. Фибраты. Прочие препараты.Что такое липиды и липопротеины?
Их значение в жизнедеятельности организма.
К липидам (жироподобным веществам) крови относятся холестерин (ХС), триглицериды и фосфолипиды. Около 700–1000 мг ХС синтезируется в организме и примерно 300–500 мг поступает с пищей. Синтез ХС осуществляется в клетках почти всех органов и тканей, однако в значительных количествах он образуется в печени – 80%, в стенке тонкой кишки – 10% и коже – 5%. ХС имеет сложное гетероциклическое стероидное ядро и выполняет следующие физиологические функции [11]. Во-первых, он является пластическим материалом, так как представляет собой обязательный структурный компонент любых клеточных мембран, обеспечивающий их стабильность. Во-вторых, из ХС в печени синтезируются желчные кислоты, которые необходимы для эмульгации и абсорбции жиров в тонком кишечнике. В-третьих, ХС является предшественником стероидных гормонов коры надпочечников (гидрокортизона и альдостерона), а также половых гормонов (эстрогенов и андрогенов).
Главным источником эндогенного ХС является печень. Основные этапы синтеза ХС представлены на схеме 1. На первом этапе этого процесса из трех молекул ацетата и коэнзима А синтезируется 3-гидрокси-3-метилглютарил коэнзим А (ГМГ-КоА). Далее в результате воздействия фермента ГМГ-КоА-редуктазы образуется мевалоновая кислота, которая примерно через 20 последующих этапов превращается в ХС. Несмотря на всю сложность и многоэтапность этих процессов, ключевым ферментом, определяющим скорость синтеза ХС, является именно ГМГ-КоА-редуктаза. Выбор этого фермента в качестве мишени для воздействия статинов позволяет решающим образом вмешиваться в синтез ХС и контролировать тем самым его уровень в плазме крови.
Схема 1.
Основные этапы синтеза холестерина
Ацетат -> ГМГ-КоА + ГМГ-КоА-редуктаза -> Мевалоновая кислота -> -> ~ 20 этапов -> -> Холестерин |
Синтезируемый в печени ХС обеспечивает потребность в нем ряда органов и тканей и, прежде всего, - самой печени, которая является не только его основным "производителем", но и "потребителем". Известно, что средний период полужизни гепатоцита составляет не более 100 дней, в связи с чем печени требуется много ХС для построения собственных клеточных мембран. Относительно небольшое количество синтезируемого ХС поступает в кровь, а основная его часть трансформируется в желчные кислоты и попадает с желчью в просвет тонкого кишечника. Из нижних отделов кишечника около 97% желчных кислот абсорбируется и возвращается в печень. Этот процесс называется энтерогепатической циркуляцией. Абсорбция желчных кислот в просвете кишечника является основным механизмом действия секвестрантов желчных кислот (анионообменных смол) - холестирамина и колестипола. Небольшие количества ХС и желчных кислот могут также связываться богатыми растительной клетчаткой пищевыми продуктами. Потребность печени в ХС удовлетворяется не только за счет его синтеза гепатоцитами, но и за счет поступления из крови. В условиях "холестеринового голода", в частности, вызванного приемом статинов, гепатоциты стимулируют специфические рецепторы, расположенные на их клеточной мембране, которые осуществляют распознавание и захват липопротеидов низкой плотности, являющихся основным холестеринсодержащим классом липопротеидов. Это рецепторы к апопротеидам В и Е (В/Е рецепторы). Активация этих рецепторов является основным условием понижения уровня ХС плазмы крови.
Триглицериды (ТГ) представляют собой эфиры трехатомного спирта глицерина и высших жирных кислот. В зависимости от количества двойных связей жирные кислоты могут быть насыщенными (нет двойных связей), мононенасыщенными (одна связь) и полиненасыщенными (две и более связи). ТГ являются важнейшим источником энергии как для скелетной муслулатуры, так и для миокарда. По своей энергетической ценности жирные кислоты вдвое превосходят глюкозу и другие моносахариды. Функция ТГ (и жирных кислот) как пластического материала заключается в их способности аккумулироваться в жировых депо. ХС и ТГ являются гидрофобными соединениями, нерастворимыми в воде и плазме крови. Они могут переноситься с током крови только в составе белково-липидных комплексов - липопротеидов (ЛП), которые представляют собой сферические частицы, имеющие электрический заряд. Наружный слой ЛП образуют белки - апопротеиды, или просто "апо", а ядро ЛП составляют липиды - ХС и ТГ. Выделяют четыре основных класса ЛП, отличающихся по размеру, удельному весу (плотности), подвижности при электрофорезе, содержанию ХС и ТГ и составу апопротеидов: хиломикроны (ХМ), ЛП очень низкой плотности (ЛПОНП), ЛП низкой плотности (ЛПНП) и ЛП высокой плотности (ЛПВП) (рис. 1).
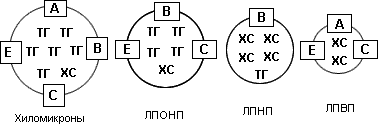
Рис. 1. Классы липопротеидов
При расположении классов ЛП в той последовательности, в которой они представлены на рис. 1 (от ХМ до ЛПВП), легко прослеживаются следующие закономерности: постепенное увеличение их плотности (наиболее "легкими" являются частицы ХМ, а наиболее "тяжелыми" - ЛПВП), усиление подвижности при электрофорезе (ХМ остаются на старте, а ЛПВП составляют наиболее мобильный класс ЛП), уменьшение размера частиц (диаметр ХМ равен примерно А, а ЛПВП - всего лишь 50-80 А), а также увеличение содержания ХС и уменьшение содержания ТГ.
Таблица 1.
Основные характеристики липопротеидов плазмы крови.
Класс ЛП | Липиды | Аполипопротеиды | Плотность, г/мл | Диаметр, А |
ХМ | ТГ>>ХС | A-I, A-II, A-IV, B-48 | <0,95 | |
ЛПОНП | ТГ>>ХС | В-100, С-I, С-II, С-III, E | <1,006 | 300-800 |
ЛППП | ТГ=ХС | В-100, Е | 1,006-1,019 | 250-350 |
ЛПНП | ХС>ТГ | В-100 | 1,019-1,063 | 180-280 |
ЛПВП | ХС>>ТГ | А-I, А-II, С-I, С-II, С-III, Е | 1,125-1,210 | 50-90 |
Хиломикроны (ХМ) – самые крупные липопротеиновые частицы. ХМ богаты триглицеридами, содержат апопротеин В-48 в качестве главного структурного белка и транспортируют экзогенные (пищевые) жиры и ХС из кишечника в печень и периферические ткани. Они образуются в эндоплазматическом ретикулуме кишечника, секретируются в лимфу и затем через грудной проток попадают в кровь. Период полужизни хиломикронов составляет 5–20 мин. Плазма крови здоровых людей, при взятии крови натощак, практически не содержит хиломикронов.
После секреции хиломикроны получают апопротеины Е, С-I, C-II и C-III от липопротеинов высокой плотности (ЛПВП). В кровотоке под действием фермента липопротеинлипазы, связанной с протеогликанами эндотелиальных клеток и активируемой с помощью апопротеина C-II, происходит гидролиз триглицеридов в составе хиломикронов. При этом хиломикроны подвергаются ремоделированию с образованием остатков (ремнант), которые имеют плотность липопротеинов очень низкой плотности (ЛПОНП), и затем – липопротеинов промежуточной плотности. Ремнанты хиломикронов, содержащие апопротеин В-48 и обогащенные апопротеином Е, захватываются гепатоцитами с помощью рецепторов, имеющих высокое сродство с апопротеином Е [11].
Параллельно печень секретирует богатые триглицеридами ЛПОНП, содержащие на поверхности молекулу апопротеина В-100. В пробах крови, взятых натощак, на долю ЛПОНП приходится около 10–15% общего ХС и практические все триглицериды крови. Апопротеин В-100, синтезируемый в печени, является составной частью не только ЛПОНП, но и ЛППП, ЛПНП, поэтому их относят к содержащим апопротеин В липопротеинам крови.
ЛПОНП являются транспортной формой эндогенных триглицеридов, на долю которых приходится около 50–70% массы частицы. Если апопротеин В-100 – интегральная часть ЛПОНП, то апопротеины Е, С-I, C-II и C-III поступают к частицам ЛПОНП уже в кровотоке от ЛПВП. Печень может секретировать как крупные, так и мелкие ЛП, богатые триглицеридами, с плотностью от липопротеинов промежуточной плотности до ЛПОНП.
Примерно половина секретированных ЛПОНП обратно захватывается печенью. Другая половина ЛПОНП после гидролиза триглицеридов в составе этих частиц под действием фермента липопротеинлипазы преобразуется в липопротеины промежуточной плотности (ЛППП). ЛППП, содержащие апопротеин Е, так же как и частицы ЛПОНП, имеют два пути метаболизма. Одна их часть удаляется из кровотока печенью с помощью рецепторов к ЛПНП. Другая же часть липопротеинов промежуточной плотности подвергается воздействию печеночной липазы, что ведет к гидролизу оставшихся триглицеридов с образованием ЛПНП. В процессе образования ЛПНП большая часть апопротеинов Е, С-I, C-II и C-III покидает ремнанты ЛПОНП и вновь ассоциируются с ЛПВП [14].
ЛПНП являются основным переносчиком эндогенного ХС в крови (транспортирует около 70% общего ХС плазмы). Его липидное ядро почти полностью состоит из эфиров ХС. Одна молекула апопротеина В-100 на поверхности ЛПНП обеспечивает распознавание, связывание и удаление около 75% частиц ЛПНП из циркуляции с помощью апо В/Е-рецепторов печени и периферических клеток. Около 3/4 ЛПНП удаляется печенью, а остальная часть – внепеченочными тканями. Период полужизни ЛПНП в крови – 2,5 дня. ЛПНП метаболизируются двумя основными путями. Первый путь метаболизма – связывание с апо В/Е-рецепторами печени, клеток надпочечников и периферических клеток, включая гладкомышечные клетки и фибробласты. В норме рецептор-опосредованным путем удаляется около 75% ЛПНП из циркуляции. После проникновения в клетку частицы ЛПНП подвергаются деградации с высвобождением свободного ХС, который выполняет регуляторную роль в метаболизме ХС – при избытке внутриклеточного ХС, через взаимодействие с геном рецептора ЛПНП, подавляет синтез рецепторов к ЛПНП. И, наоборот, при низком уровне внутриклеточного ХС синтез рецепторов к ЛПНП возрастает.
Альтернативный путь катаболизма частиц ЛПНП – окисление. Перекисно-модифицированные ЛПНП, образовавшиеся в результате воздействия эндотелиальных клеток, гладкомышечных клеток или моноцитов/макрофагов слабо распознаются апо В/Е-рецепторами, но быстро распознаются и захватываются так называемыми скэвенджер (в переводе с англ. scavenger – мусорщик) - рецепторами макрофагов. Этот путь катаболизма ЛПНП в отличие от рецептор-зависимого пути, не подавляется при увеличении количества внутриклеточного ХС. Продолжение этого процесса приводит к превращению макрофагов в переполненные эфирами ХС пенистые клетки – компоненты жировых пятен. Последние являются предшественниками атеросклеротической бляшки [1, 11].
Прямое определение концентрации ЛПНП в крови – дорогостоящая и трудоемкая задача. В большинстве случаев определяют содержание общего ХС, триглицеридов и части ХС, транспортируемой ЛПВП, а концентрацию ХС ЛПНП рассчитывают по формуле Friedwald [18]:
ХС ЛПНП, ммоль/л = общий ХС – ХС ЛПВП – (0,45 х триглицериды)
ХС ЛПНП, мг/дл = Общий ХС – ХС ЛПВП – (0,2 х триглицериды)
Расчет ХС ЛПНП по формуле Friedwald правомерен в случае, когда концентрация триглицеридов менее 5 ммоль/л (450 мг/дл).
ЛПВП – самые мелкие липопротеиновые частицы. На их долю приходится 20–30% общего ХС крови, но из всех липопротеинов именно эти частицы содержат наибольшее количество фосфолипидов и белка. ЛПВП образуются в печени и кишечнике в виде незрелых дисковидных частиц, состоящих из фосфолипидов, апопротеинов семейства А (А-I и А-II) и ХС. Еще один источник ЛПВП – это преобразование липопротеинов и апопротеинов в процессе метаболизма и ремоделирование богатых триглицеридами частиц – хиломикронов и ЛПОНП.
Основная функция ЛПВП в обмене липопротеинов – обеспечение обратного транспорта ХС. Обратный транспорт ХС – позитивный процесс, с помощью которого ХС возвращается из периферических тканей в печень для дальнейшего катаболизма. По современным представлениям, незрелые частицы ЛПВП – хорошие акцепторы свободного ХС. Свободный ХС на поверхности ЛПВП эстерифицируется с образованием эфиров ХС. В роли катализатора эстерификации свободного ХС выступает фермент лецитин-холестерин-ацетилтрансфераза, а в качестве кофактора – апопротеин А-1, структурный белок ЛПВП. Образованные эфиры ХС перемещаются с поверхности частиц ЛПВП в гидрофобное ядро, освобождая таким образом дополнительную поверхность для свободного ХС. По мере накопления в ядре эфиров ХС, дисковидные частицы ЛПВП преобразуются в сферические, богатые холестерином ЛПВП. Эфиры ХС из ЛПВП и содержащих апопротеин В липопротеинов захватываются гепатоцитами через рецептор-опосредованный эндоцитоз или с помощью скэвенджер-рецепторов.
За сутки в организме человека окисляется около 500 мг ХС в желчные кислоты, примерно такое же количество выделяется с фекалиями и около 100 г – с кожным жиром. Свободный, неэстерифицированный ХС содержится в мембранах клеток. Мозг, желчь и эритроциты содержат только свободный ХС, скелетная мышца и надпочечники – и свободный, и эстерифицированный ХС.
Помимо описанных 5 классов ЛП выделяют ЛП (а). В структурном отношении они идентичны ЛПНП, но содержат дополнительный апо-протеин - апо (а), связанный дисульфидным мостиком с апо В-100. Показано, что ЛП (а) является независимым фактором риска ИБС. Атерогенные окисленные формы ЛП (а) образуются значительно легче, чем окисленные формы ЛПНП. В связи со структурным сходством с плазминогеном ЛП (а) рассматриваются как конкурентные антагонисты плазминогена, ассоциирующиеся с повышенным риском возникновения тромбоза коронарных артерий [1, 14].
Атерогенные и неатерогенные липопротеины
Липопротеины различаются и по участию в атерогенезе. Атерогенность липопротеинов частично зависит от размера частиц. Самые мелкие липопротеины, такие как ЛПВП, легко проникают в стенку сосуда, но также легко ее покидают, не вызывая атеросклероз. Богатые триглицеридами частицы – хиломикроны и крупные ЛПОНП, как полагают, не атерогенны, но их избыток может вызвать острый панкреатит. Что касается остатков липолиза богатых триглицеридами липопротеинов – ремнант хиломикронов и липопротеинов промежуточной плотности, то они считаются атерогенными. ЛПНП, липопротеины промежуточной плотности и мелкие ЛПОНП достаточно малы, и в случае химической модификации (вследствие окисления) легко задерживаются в сосудистой стенке.
ЛПНП – наиболее атерогенные липопротеины крови. Строгая, независимая прямая связь между уровнем ХС ЛПНП и риском ИБС четко установлена как у мужчин, так и у женщин, у лиц без признаков ИБС и больных ИБС. По расчетам, увеличение концентрации ХС ЛПНП на 1% может привести к увеличению риска ИБС на 2–3 %.
Согласно современным рекомендациям снижение концентрации ХС ЛПНП – стратегическая цель профилактики ИБС и основная задача диетического и медикаментозного вмешательства при дислипидемии [8]. Помимо уровня ХС ЛПНП, на риск возникновения ИБС влияет и размер частиц ЛПНП. По данным проспективных исследований, у людей, у которых в крови преобладают мелкие, плотные частицы ЛПНП (фенотип В), риск ИБС более чем в 3 раза выше, независимо от уровня ХС ЛПНП [18].
Единое мнение об истинном значении триглицеридов в отношении увеличения риска ИБС пока отсутствует. При одномерном анализе большинства проспективных исследований уровень триглицеридов до 5 ммоль/л (450 мг/дл) предсказывает риск ИБС, особенно у женщин. Так, в Фрамингемском исследовании риск ИБС был тем выше, чем выше была концентрация триглицеридов. Но когда вводится поправка на другие факторы риска, особенно ЛПВП, независимый эффект триглицеридов исчезает или значительно ослабевает. Однако в последнее время стали появляться факты, свидетельствующие о независимой связи концентрации триглицеридов с риском ИБС [27, 34].
Одна из главных причин, затрудняющих оценку высокого уровня триглицеридов как независимого фактора ИБС – это гетерогенность липопротеинов, богатых триглицеридами, содержащих апопротеин В. По мнению Brewer (1999), также как существует “хороший и плохой” ХС (ХС ЛПВП и ХС ЛПНП), есть два вида гипертриглицеридемии. Некоторые случаи гипертриглицеридемии ассоциируются с высоким риском ИБС, а другие – нет. Например, выраженная гипертриглицеридемия может быть за счет хиломикронов и крупных частиц ЛПОНП, однако они слишком крупные и не могут проникнуть в стенку сосуда. Синдром семейной хиломикронемии, в основе которого лежит дефицит либо липопротеинлипазы – фермента, ответственного за гидролиз триглицеридов, либо апопротеина С-II (кофактора фермента липопротеинлипазы), ассоциируется с низким риском ИБС, но повышенной вероятностью развития острого панкреатита. Также с низким риском ИБС ассоциируется гипертриглицеридемия, вызванная злоупотреблением алкоголя, приемом эстрогенов и некоторыми семейными формами гипертриглицеридемии. В отличие от крупных частиц ЛПОНП мелкие формы ЛПОНП, так же как и липопротеины промежуточной плотности, – атерогенны, и лица с гипертриглицеридемией за счет высокой концентрации мелких ЛПОНП и липопротеинов промежуточной плотности имеют высокий риск ИБС.
Тесная обратная связь между уровнем ХС ЛПВП и риском ИБС обнаружена у мужчин и женщин, у лиц без клинических проявлений атеросклероза и больных ИБС. По данным результатов 4-х больших эпидемиологических исследований (Framingam, LRS Prevention Mortality-Follow up Study, MRFIT), сделано заключение, что повышение ХС ЛПВП на 1 мг/дл (0,026 ммоль/л) сопряжено со снижением риска сердечно-сосудистых заболеваний на 1,9-2,9 % [18]. Механизм обратной взаимосвязи между ХС ЛПВП и ИБС не вполне ясен. ХС ЛПВП обычно снижен, когда уровень триглицеридов высок, и допускается вероятность того, что концентрация ХС ЛПВП – это лишь реципроктное отражение уровня атерогенных ЛП, таких как ЛПОНП. Не исключается возможность прямого защитного действия ЛПВП на артериальную стенку с помощью транспорта ХС из артериальной стенки в печень или ингибирования окисления ЛПНП.
Отрицательный совокупный эффект различных липидов и липопротеинов плазмы очень важен, так как сочетание гипертриглицеридемии с низким ХС ЛПВП и отношением общий ХС/ХС ЛПВП>5 связано особенно с высоким риском ИБС. Так, например, группа мужчин и женщин – участников Фрамингемского исследования с уровнем триглицеридов >150 мг/дл и уровнем ХС ЛПВП<50 мг/дл имела самую высокую частоту возникновения ИБС и ее нельзя было идентифицировать по уровню ХС ЛПНП, который в этой группе был в пределах 120–125 мг/дл.
Первичные и вторичные дислипидемии.
При дислипидемии концентрация липидов и липопротеинов крови выходит за пределы нормы вследствие наследственных или приобретенных состояний, при которых нарушается их образование, разрушение или удаление из циркуляции. Дислипидемии классифицируются в зависимости от того, уровень каких именно липидов и липопротеинов выходит за пределы нормы.
Одна из первых классификаций гиперлипидемий принадлежит Фредриксону (1967), который, скомбинировав результаты нескольких методов разделения липопротеинов: электрофореза на бумаге, преципитации с гепаринсульфатом и препаративного ультрацентрифугирования, предложил выделять 5 фенотипов гиперлипидемий (табл. 2). К ее недостаткам следует отнести то, что она не разделяет первичные и вторичные дислипидемии, не учитывает уровень ХС ЛПВП и генетические дефекты, лежащие в основе многих нарушений липидного обмена.
Таблица 2.
Классификация первичных гиперлипидемий по Фредриксену (1967 г.).
Фенотипы | Липопротеины, содержание которых увеличено | Уровень ХС | Уровень триглицеридов | Атерогенность |
I | Хиломикроны | В норме или ↑ | ↑↑↑↑ | Не установлено |
II а | ЛПНП | ↑↑ | В норме | +++ |
II б | ЛПНП и ЛПОНП | ↑↑ | ↑↑ | +++ |
III | ЛППП | ↑↑ | ↑↑↑ | +++ |
IV | ЛПОНП | В норме или ↑ | ↑↑ | + |
V | ЛПОНП и хиломикроны | ↑↑ | ↑↑↑↑ | + |
Первичные дислипидемии, характеризующиеся гиперхолестеринемией.
Многие фенотипы гиперлипидемий генетически детерминированы. Причиной высокого уровня общего ХС часто является семейная гиперхолестеринемия – моногенное нарушение, вызванное мутацией гена ЛПНП-рецепторов. Частота гетерозиготных форм этого нарушения в большинстве популяций составляет 1 на 500. Обычно от каждого родителя наследуется один ген рецептора ЛПНП. При гетерозиготной форме семейной гиперхолестеринемии у больного имеется только один нормальный ген рецептора ЛПНП, уровень ХС ЛПНП превышает 200 мг/дл, общего ХС плазмы – 300 мг/дл, встречаются ксантомы сухожилий, липоидная дуга роговицы и преждевременно развивается ИБС. На долю гетерозиготной семейной гиперхолестеринемии приходится до 2–5% всех случаев ИБС у лиц в возрасте до 60 лет. [1]
Редко, в одном случае на миллион встречаются лица, наследующие оба ненормальных гена рецептора ЛПНП, и они, следовательно, являются гомозиготными по признаку семейной гиперхолестеринемии. Уровень ХС у таких больных колеблется в пределах 15,5– 25,9 ммоль/л (600–1000 мг/дл), у них наблюдаются плоские и эруптивные ксантомы сухожилий. Тяжелая и подчас летальная форма коронарной недостаточности развивается уже к 13-19 годам.
Еще одним примером моногенных дислипидемий является семейный дефект апопротеина В (familial defective apo B), вызванный мутацией гена апопротеина В, встречается с частотой 1:500 и тоже сопровождается увеличением концентрации ХС и ХС ЛПНП и высоким риском развития ИБС. Высокие уровни ХС и ХС ЛПНП у пациента с семейным анамнезом преждевременной ИБС должны насторожить относительно наличия семейной гиперхолестеринемии, они являются показанием для исследования концентрации липидов и липопротеинов у близких родственников (родители, дети, братья и сестры).
Наиболее частой причиной изолированной гиперхолестеринемии (IIa тип гиперлипидемии) является полигенная гиперхолестеринемия. Концентрация общего ХС и ХС ЛПНП при полигенной семейной гиперхолестеринемии меньше, чем при гетерозиготной семейной гиперхолестеринемии: у больного отсутствуют ксантомы сухожилий, но преждевременное возникновение ИБС – характерное явление.
Семейная комбинированная гиперлипидемия может проявиться в виде IIб или IV типа гиперлипидемии. Ключевым нарушением является увеличение синтеза апопротеина В-100 печенью, что в свою очередь сопровождается увеличением количества богатых триглицеридами липопротеинов в плазме. При IIб типе содержание общего ХС достигает 250–350 мг/дл, при IV типе – отмечается умеренная гипертриглицеридемия, но может быть и существенный рост концентрации триглицеридов. Встречается в популяции с частотой 1 на 100. У больных отсутствуют ксантомы сухожилий, а ИБС возникает в зрелом возрасте.
Первичные дислипидемии с гипертриглицеридемией.
Семейная хиломикронемия – редкая наследственная патология, которая характеризуется присутствием хиломикронов в плазме крови, взятой натощак (V тип гиперлипидемии). Содержание триглицеридов в крови резко увеличено, концентрация ХС нормальная или слегка повышена. В основе лежит обусловленное генетическими нарушениями снижение активности фермента липопротеинлипазы или его кофактора апопротеина С-II. Больных беспокоят боли в животе, присутствуют эруптивные ксантомы, развивается сопутствующий панкреатит, но риск ИБС не увеличивается.
Дисбеталипопротеинемия, или III тип гиперлипидемии, характеризуется увеличением количества липопротеинов промежуточной плотности, что проявляется гиперхолестеринемией, гипертриглицеридемией и высокой вероятностью раннего развития ИБС. Встречается с частотой 1 на 5000. В основе дисбеталипопротеинемии лежит полиморфизм гена апопротеина Е. Нормальный фенотип апопротеина Е обозначается как Е-3, а III тип гиперлипидемии вызван наличием изоформы апопротеина Е-2, который эффективно не связывается с апо В/Е-рецепторами клеток и рецепторами к ремнантам, что ведет к нарушению удаления липопротеинов промежуточной плотности и их накапливанию. Липопротеины промежуточной плотности при дисбеталипопротеинемии обладают бета-подвижностью при электрофорезе, обогащены эфирами ХС, захватываются макрофагами, и поскольку этот путь катаболизма не регулируется по механизму обратной связи уровнем внутриклеточного ХС, приводит к превращению макрофагов в пенистые клетки.
Дисбеталипопротеинемия наиболее часто проявляется у гомозигот по апопротеину Е-2. Диагностируется дисбеталипопротеинемия с помощью выявления изоформы апопротеина Е, но величина отношения ХС ЛПОНП/триглицериды плазмы более 0,3 подтверждает диагноз дисбеталипопротеинемии.
Еще одним типом семейной гипертриглицеридемии является семейная эндогенная гипертриглицеридемия (IV тип гиперлипидемии), для которой характерно увеличение содержания в плазме ЛПОНП. Уровень триглицеридов находится в пределах 200-500 мг/дл, концентрация ХС ЛПВП снижена, общего ХС – в норме или умеренно повышена, встречается с частотой 1 на 300 и в ряде случаев приводит к раннему возникновению ИБС [14].
Вторичные дислипидемии.
Перечень заболеваний, синдромов и факторов, пиводящих к развитию вторичных гиперлипидемий, а также характер отмечающихся при этом нарушений обмена липидов [11], представлены в таблице 3.
Таблица 3.
Вторичные дислипидемии.
Заболевания, синдромы, факторы и состояния | Характер липидных |
Сахарный диабет | ТГ ↑, ХС ЛПВП ¯ |
Гипотиреоз | ОХС ↑ |
Алкоголь | ТГ ↑ |
Лекарственные препараты (диуретики, контрацептивы, анаболики, прогестагены) | ТГ ↑, ХС ЛПВП ¯, ОХС ↑ |
Хроническая почечная недостаточность | ТГ ↑ |
Нефротический синдром | ОХС ↑, ТГ ↑ |
Холестаз | ОХС ↑ |
Булимия | ТГ ↑ |
Беременность | ТГ ↑ |
Диагностика и концепция развития метаболического синдрома.
Впервые в 1923 году шведский врач Kylin описал синдром, получивший название "гипертензия-гипергликемия-гиперурикемия". А в 1960 году Sinith в своей монографии "Инсулин и атерома" выдвинул гипотезу о роли инсулина в развитии атеросклероза и связанных с ним заболеваний. К концу 80-х годов завершились наиболее крупные эпидемиологические работы, проводимые с целью определения факторов риска сердечно-сосудистых заболеваний и их осложнений: Фрамингемское исследование и Парижское проспективное исследование. Результаты этих исследований показали, что повышенная концентрация инсулина (гиперинсулинемия) прогнозировала развитие артериальной гипертонии, СД 2 типа и ИБС, являясь независимым фактором атеросклероза. Показано, что наряду с курением, артериальной гипертонией и гиперхолестеринемией, СД 2 типа, гиперинсулинемия, ожирение, повышенный уровень фибриногена в крови также являются основными факторами риска развития ИБС. Обобщая эти данные, американский ученый G. Reaven в 1988 году выдвинул гипотезу о так называемом метаболическом синдроме или синдроме X, в основе которого лежит сочетание артериальной гипертонии, гипертриглицеридемии, сопровождающейся сниженным уровнем ХС ЛВП, гиперинсулинемии, НТГ или диагностированного СД 2 типа [43]. Reaven не отнес абдоминальное ожирение к числу обязательных признаков синдрома. Однако уже в 1989 году J. Kaplan описал «смертельный квартет», включив в него абдоминальное ожирение [38].

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 |
