

Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Известно, что азот накапливается в организме млекопитающих в виде аммиака, который чрезвычайно ядовит даже в незначительных количествах. Рыбы, имея большое количtство воды, выводят его в виде очень разбавленного раствора. Наземные животные должны экономить воду, поэтому они вынуждены были превращать аммиак в менее ядовитую мочевину, а вот птицы и пресмыкающие из-за недостатка воды переводят аммиак в совершенно нерастворимую мочевую кислоту, которая совершенно безвредна для них и выделяется в виде небольших твёрдых конкреций. Такова иллюстрация естественного отбора и единства биохимического состава, строения всех живых форм, населяющих в настоящее время Землю. Формы жизни изменялись в процессе эволюции в течении многих миллионов лет. И хотя они в ходе этого процесса разошлись по множеству разнообразных направлений с изменением внешних форм, их биохимическое содержание (органическая составляющая и влияние воды) сохранилось с поразительным постоянством. Это можно объяснить тем, что биохимические свойства установились и закрепились прежде, чем виды в процессе эволюции разошлись по известным биологическим направлениям. Поскольку биохимические формы одинаковы для всех организмов, то можно предположить наличие одного предка, биохимия которого была близкой к бихимии современных живых существ. Это взаимоотношение можно представить на схеме.
Первоначальный живой организм:
Вирусы.
Бактерии.
Растения
Человек
Образование живой клетки сложный химический процесс. В течение многих лет основная масса человечества безоговорчно утверждала, что всё живое произошло только из живого. Но в определённых условиях на первобытной Земле под воздейтвием солнечной энергии начался синтез органических молекул, затем аминокислот, которые под воздействием катализаторов полимеризовалсь в крупные органические молекулы. Последние впоследствие образовали коацерваты, внутри которых проиходили свои химические процессы. Эти формы, изменяясь в процессе эволюции в течение многих миллиардов лет, и создали живую клетку- прародительницу сегоднейшей жизни.
Влияние растворителя на реакционную способность
амбидентных анионов.
Влияние растворителей на протекание химических реакций огромно. Вода, метанол и формамид сильно сольватируют и катионы и анионы. Ацетонитрил плохо сольватирует все ионы. Димелформамид, диметилацетамид, диметилсульфоксид и гексаметилфосфотриамид сильно сольватируют катионы и плохо сольватируют анионы. Это можно продемонстрировать на примере амбидентных ионов, т. е. молекул, содержащих как минимум два нуклеофильных центра. Такими центрами являются, например: углерод и кислород в фенолах и нафтолах; азот и кислород в нитритах; азот и сера в изотиоцанатах; фосфор и кислород в фосфитах; сера и кислород в тиофосфорных кислотах и т. д. Замена растворителя изменяет состав продуктов реакции. Так, увеличение сольватирующей силы растворителя промотирует реакцию по менее сольватируемому центру.
Например, фенолят-ионы взаимодействуют с аллилгалогенидами в спирте с образованием как О-, так и С-алкилированных продуктов [39]. При переходе от спирта к воде повышается выход С-алкилированного продукта.
С6Н5О - +СICH2-CH=CH2 этанол® C6H5OCH2CH=CH2
С6Н5О - +СICH2CH=CH2 вода ® С6Н4(ОН)СН2СН=СН2
При реакции b-нафтолята натрия с бромистым бензилом в ДМФ или ДМSO С-алкилирования не наблюдается, однако оно составляет 22% при проведении реакции в диметиловом эфире этиленгликоля, 36%- в ТГФ, 34% в метаноле, 28%- в этаноле, 85% - в воде. Состав продуктов реакции практически не зависит от катиона алкоголята в спиртах, но в диэтиловом эфире, в ТГФ, бензоле доля С-алкилированного продукта умеьшается в следующей последовательности: литий, натрий, калий и тетраалкиламмоний. Эти данные показывают, что когда фенолят-ион сильно сольватирован или связан с небольшим катионом в прочную ионную пару, то он реагирует преимущественно по углеродному атому. Аналогично протекают реакции с другими амбидентными ионами: нитритами, роданидами, фосфитами, тиофосфатами и т. д.[40].
Растворители с основными и кислотными функциональными группами, которые содержат атомы кислорода, широко используются как экстрагенты в аналитической химии. Наибольшей селективностью при экстракции катионов обладают фосфорорганические кислоты. Например, ди-2-этилгексилортофосфорная кислота обеспечивает возможность разделения близких по химическим свойствам лантаноидов и актиноидов, урана и тория, а трибутилфосфат в кислых растворах помогает разделить золото и тяжёлые металлы. Для экстракции кислых продуктов получили распространение азот - и серусодержащие экстрагенты типа триоктиламина, тиоэфиров, хелатообразующие и макроциклические экстрагенты.[41].
СТЕРИЧЕСКИЕ АСПЕКТЫ ОБРАЗОВАНИЯ ВОДОРОДНЫХ СВЯЗЕЙ
В образовании водородных связей определённую роль играют стерические эффекты заместителей в молекулах акцепторов и доноров водородной связи или эффекты экранирования, так как константа равновесия в водных растворах определяется не только энтальпией, но и энтропией. Так, ди-о-трет-бутилфенол практически не ассоциирован и образует с тетрагидрофураном слабую водородную связь[42]. Третичный бутиловый спирт также образует слабые Н-связи, его кислотность несколько ниже, чем у воды и метанола. Возможно, что это связано с затруднениями при сольватации трет-бутил-аниона, поскольку атом кислорода сильно заслонён метильными группами и молекулам воды достигнуть его сложнее. [42]. Некоторые красители, содержащие феноксильную группу, к примеру бромфталеиновый красный очень сильно меняет свой электронный спектр от жёлтого ( l 4000А° ) в красный с триэтиламином (l 5400А° ), а с вторичными аминами до синего ( l 5800 А° ). Предполагают, что соли с вторичными аммониевыми катионами имеют димерную циклическую структуру.[43]. Стерические эффекты достаточно подробно исследованы на примерах разветвлённых спиртов, аминов, орто - замещённых фенолов в работах Чартона, Гаммета, Тафта, Пальма. [43].
ЗНАЧИМОСТЬ ВОДОРОДНОЙ СВЯЗИ. РАСТВОРИМОСТЬ
Водородные связи, представляющие одну из форм слабых химических взаимодействий, играют существенную роль в химии водных растворов. Вода является прекрасным амфотерным растворителем. Из-за водородных связей она способна к образованию ассоциатов и имеет высокую диэлектрическую проницаемость. В воде растворяется большинство неорганических и органических кислот, оснований, солей. Из ковалентных водородных соединений в воде хорошо растворяются те, которые подвергаются электролитической ионизации с образованием гидратированных ионов ( НСI, НСIО и др.), так сказать, микроскопические айсберги вокруг ионов растворённого вещкства. NaCI, KNO3, растворяясь в воде, охлаждают её за счёт образования гидратированных ионов. Гидроокиси щелочных металлов очень хорошо растворимы в воде, причём растворение сопровождается значительным выделением тепла ( до 10ккал/моль).
Некоторые вещества способны давать межмолекулярные водородные связи с молекулами воды, например, такие как NH3, H2NNH2 , спирты и т. д. Из органических веществ растворимы в воде те, молекулы которых содержат полярные группы: многие кислоты, спирты, амины, сахара и т. д.
![]()
![]() Такие сильные неорганические кислоты, как азотная, серная, хлорная нацело диссоциируют в водных растворах. Рассмотрим кратко их некоторые свойства и водородные связи. В ряду кислородных кислот хлора с увеличением числа атомов кислорода возрастает сила кислоты, её прочность, растворимость, возможность образовывать водородные связи, а с другой стороны в этих кислотах уменьшается окислительная активность. Это обусловлено особенностями химического строения оксоанионов хлора, так как при увеличении степени окисления хлора электронные облака всё больше оттянуты к центральному атому и связь О - Н делается всё более ионной, что ведёт к усилению кислотных свойств. Эти кислоты можно представить следующими формулами: H-O-CI, H-O-CI=O, H-O-CIO2, H-O-CIO3. В солях их можно представить в виде ионов: [ О =СI=O ]-.
Такие сильные неорганические кислоты, как азотная, серная, хлорная нацело диссоциируют в водных растворах. Рассмотрим кратко их некоторые свойства и водородные связи. В ряду кислородных кислот хлора с увеличением числа атомов кислорода возрастает сила кислоты, её прочность, растворимость, возможность образовывать водородные связи, а с другой стороны в этих кислотах уменьшается окислительная активность. Это обусловлено особенностями химического строения оксоанионов хлора, так как при увеличении степени окисления хлора электронные облака всё больше оттянуты к центральному атому и связь О - Н делается всё более ионной, что ведёт к усилению кислотных свойств. Эти кислоты можно представить следующими формулами: H-O-CI, H-O-CI=O, H-O-CIO2, H-O-CIO3. В солях их можно представить в виде ионов: [ О =СI=O ]-.
О О
В отличие от кислородных кислот хлора молекула НСI малополярна и водородные связи очень слабы. Вследствие этого растворимость НСI в воде ограничена - 24-38%. При растворении НСI в воде имеет место протолитическая реакция: НСI + Н2О ® Н3+О +СI - и образуется сильная соляная кислота (рКa= -7).
Фтористый водород, несмотря на ионность своих молекул, смешивается с водой во всех пропорциях, образуя среднюю по силе плавиковую кислоту в виде цепочечных или циклических ассоциатов. Аномальное поведение её объясняется большой энергией водородной связи между молекулами НF - 83 кДж/моль, что затрудняет гетеролитический распад НF в воде, который также осложняется образованием устойчивых гидрофторид – анионов типа (НF2-,, Н2 F3-).[ 44].
 Безводная серная кислота, называемая также моногидратом, является сильно ассоциированным растворителем. Исследования показывают, что она имеет особую структуру, в которой каждая молекула Н2SО4 связана посредством водородной связи с четырьмя другими такими же молекулами. Серная кислота как растворитель имеет высокую диэлектрическую проницаемость (e= 100,5), самую высокую константу самоионизации Кs = 2,7 10-4 и способна отнимать гидратную воду у веществ. При смешивании её с водой выделяется значительное количество тепла (20.42 ккал/моль). В водных растворах ведёт себя как сильнейшая двухосновная кислота ( рК1 =3 и рК2 =2), а ион - SO42- имеет форму правильного тетраэдра; все расстояния и углы между связями в нём равноценны:
Безводная серная кислота, называемая также моногидратом, является сильно ассоциированным растворителем. Исследования показывают, что она имеет особую структуру, в которой каждая молекула Н2SО4 связана посредством водородной связи с четырьмя другими такими же молекулами. Серная кислота как растворитель имеет высокую диэлектрическую проницаемость (e= 100,5), самую высокую константу самоионизации Кs = 2,7 10-4 и способна отнимать гидратную воду у веществ. При смешивании её с водой выделяется значительное количество тепла (20.42 ккал/моль). В водных растворах ведёт себя как сильнейшая двухосновная кислота ( рК1 =3 и рК2 =2), а ион - SO42- имеет форму правильного тетраэдра; все расстояния и углы между связями в нём равноценны:
![]()
![]()
![]() O=S-OH и [ O =S=O]-2
O=S-OH и [ O =S=O]-2
O OH O
![]()
![]() Азотная кислота считается одной из сильнейших кислот, но в безводной серной кислоте ведёт себя как основание: НNO3 + Н2SO4 D H2NO3+ + НSО4- Молекула НNO3 и нитрат - ион имеют строение, представленное схемами: O=N=O « [ O= N= O ]- + H+
Азотная кислота считается одной из сильнейших кислот, но в безводной серной кислоте ведёт себя как основание: НNO3 + Н2SO4 D H2NO3+ + НSО4- Молекула НNO3 и нитрат - ион имеют строение, представленное схемами: O=N=O « [ O= N= O ]- + H+
ОH O
Фосфорная кислота Н3РО4- одно из наиболее важных многочисленных производных пятивалентного фосфора. В твёрдой кислоте и концентрированных растворах действуют межмолекулярные водородные связи. Фосфорная кислота смешивается с водой во всех соотношениях. В более разбавленных растворах возникают водородные связи между молекулами воды и кислоты. По химическому строению она представляет собой искажённый тетраэдр, в котором три вершины заняты гидроксильными группами, а четвёртая - атомом кислорода. В центре тетраэдра находится атом фосфора с sр3 гибридными орбиталями. В водной среде Н3РО4- кислота средней силы: рК1 2,2; рК2 7,3; рК3 12.4 и это позволяет ей поддерживать во многих реакциях нейтральную буферную среду. Фосфорная кислота играет важную роль в биохимических процессах. Широко известны фосфаты сахаров, аминокислот, оксикислот. Кроме того, фосфорная кислота и её ионы обладают способностью конденсироваться, образовывая ди - и трифосфаты. Последние принимают активное участие в образовании нуклеотидов, в частности, аденозинтрифосфата ( АТФ ), который является главным носителем химической энергии в клетках всех живых организмов. АТФ образуется в клетке многими путями: при внутриклеточном расщеплении глюкозы, при разложении жирных кислот и аминокислот. При передаче своей энергии другим молекулам АТФ теряет концевую фосфатную группу и превращается в аденозиндифосфат (АДФ). При этом выделяется 8000 кал. АДФ в процессе фосфорилирования либо за счёт фотосинтеза в растениях, либо химической энергии при питании органическими веществами, может вновь превратится в АТФ. Каждая фаза клеточной деятельности характеризуется участием этого универсального переносчика энергии. Дополнительным аккумулятором энергии является и креатинфосфат (КрФ), при гидролизе которого образуется креатин, неорганический фосфат и около 8000 кал. Без этих нуклеотидов клетка истощается и утрачивает способность исполнять свои физиологические функции.[45].
Фосфорная кислота входит в основные цепи ДНК и РНК- молекулы наследственности. Последние представляют собой двухнитевые молекулы, прочность которых обусловлена кооперативными водородными связями. Можно предположить, что и на других планетах фосфорная кислота и её соли при отсуствии воды могли бы стать её заменителями.
Фосфористая кислота Н3РО3 – это хорошо растворимые в воде кристаллы, образует водородные связи. Это кислота средней силы: рК 2,8 ; рК 6,2. Она способна окисляться многими окислителями, в том числе и галогенами, превращаясь в фосфорную кислоту:
![]() (НО)2 Р=О + СI2 + Н2О → Н3РО4 + 2 НСI
(НО)2 Р=О + СI2 + Н2О → Н3РО4 + 2 НСI
Н
РАСТВОРИМОСТЬ АЗОТСОДЕРЖАЩИХ ВЕЩЕСТВ В ВОДЕ
Электроотрицательность атома азота больше, чем фосфора, поэтому связь N-Н является более поляризованной, чем связь Р-Н, и в жидком аммиаке между его молекулами действуют сильные водородные связи, придавая ему ряд экстремальных свойств. Жидкий аммиак является прекрасным растворителем для щелочных металлов, электрона, солей, кислот. Несмотря на минусовые температуры в нём могут протекать ряд удивительных реакций, связанных, например, с алкилированием металлических производных ацетилена[46], реакций обмена, аммонирования, комлексообразования, окисления-восстановления.[ 38].
Вследствие полярности и достаточно высокой диэлектрической проницаемости молекул газообразного аммиака он превосходит по растворимости многие газы: при О°С один объём воды поглощает 1200 объёмов газообразного аммиака. Прекрасная растворимость аммиака в воде обусловлена возникновением межмолекулярных водородных связей. При этом теоретически возможны два механизма возникновения связи между молекулами аммиака и воды:
Н3 N: …Н-ОН и H2N-Н…ОН2
Большая электроотрицательноть атома кислорода по сравнению с азотом приводит к тому, что атом кислорода более прочно удерживает электроны, чем атом азота. Вследствие этого, неподелённая пара электронов атома азота легче образует Н- связь, чем неподелённая пара кислорода, да и донорские способности молекулы аммиака выражены сильней, чем у воды. Поэтому межмолекулярная водородная связь образуется по первому механизму. Физико-химические процессы в водном растворе аммиака можно представить двумя константами равновесия по схеме:
Кравн= 0,2 Кравн= 10-5
NН3 + Н2ОD NН3 + ОН2 D NН4+ + ОН-
Возникновение гидроксил - ионов создаёт щелочную среду в аммиачной воде. Равновесие в водном растворе аммиака можно сместить вправо добавлением кислоты, в растворе образуются соли аммония, которые прекрасно растворяются в воде.[47]. Аналогично реагируют с водой другие амины, например, триэтиламин, пиридин:
С5Н5N + HOH®C5H5N+H + OH-
КЕТО-ЕНОЛЬНОЕ РАВНОВЕСИЕ
Многие енольные формы карбонильных соединений стабилизируются образованием водородных связей. Ацетон в равновесии содержит только 0,00025% енола, т. е. равновесие практически нацело сдвинуто влево:
СН3 С=ОСН3 D СН3 С(ОН)=СН2
Альдегиды легче енолизируются, чем кетоны, так как разница энергий образования альдегидной и енольной формы у них несколько меньше. Кето-енольное равновесие зависит от наличия различных группировок по соседству с карбонильной группой. Циклические кетоны сильнее енолизированы, чем алифатические. Содержание енола в ацетоне - 0,00025%, в циклогексаноне - 0,020%, в диацетиле-0,0056%, а циклогександион–1,2 енолизирован на 100%. Это объясняется двумя факторами: дипольным отталкиванием кетонных групп в жёстком гексановом кольце и внутримолекулярной водородной связью.
В ацетилацетоне енол составляет 80%, так как он стабилизирован внутримолекулярной водородной связью:
![]()
![]()
![]()
![]() СН2 –СН2 СН =СОН
СН2 –СН2 СН =СОН
![]()
![]() СН2 С=О D СН2 СН2
СН2 С=О D СН2 СН2
![]()
![]()
![]() СН2 - СН2 СН3 СН2- СН2
СН2 - СН2 СН3 СН2- СН2
![]()
![]()
![]()
![]() O=С-СН2- С=ODO=С-СН= С-СН3
O=С-СН2- С=ODO=С-СН= С-СН3
CH3 СН3 Н-О
Введение электроотрицательных радикалов ( СI, СN, С6Н5 ) к кетонной группе увеличивает содержание енольной структуры. При замене метильной группы на фенильную наблюдается возрастание содержания енола до 99 %. Замена её на алкоксильный радикал уменьшает енолизацию. Ацетоуксусный эфир содержит лишь 7,5% енола, что объясняется более слабыми электроноакцепторными свойствами сложноэфирной группировки по сравнению с кетонной. При замене карбэтоксильной группы альдегидной получаются b-кетоальдегиды, в которых равновесие сильно смещено в сторону енольной формы, при этом енолизация происходит за счёт альдегидной группы:
![]()
![]() O=C-CH2C=O D O=C-CH=CH-OH
O=C-CH2C=O D O=C-CH=CH-OH
![]() R Н R
R Н R
При замене метильных групп в ацетилацетоне на трет-бутильную возрастает процент енольной формы в 4-5 раз, что обяъясняют стерическими напряжениями в дикетонных формах. Циклопентандионы-1,3 практически не енолизированы, а циклогександионы-1,3, напротив, енолизированы значительно за счёт образования димеров с водородными связями.[48,49]
Константы енолизации многих органических соединений зависят от скорости отрыва протона в том или ином растворителе. Если заменить воду более сильным основанием, то скорости увеличатся. в ряде работ вывел интересные соотношения, характеризующие кето-енольное равновесие.[50].
Метаболизм живой клетки постоянно использует кето-енольное равновесие. Так, анаэробное окисление глюкозы в пировиноградную кислоту происходит через ряд стадий, каждую из которых катализирует специальный фермент. Образующийся на одной из стадий ненасыщенный богатый энергией фосфат- енол, превращает АДФ в АТФ:
![]()
![]()
![]() СН2=С - О - РО3Н2 + АДФgСН2=С-ОН +АТФ g СН3-С=О +АТФ
СН2=С - О - РО3Н2 + АДФgСН2=С-ОН +АТФ g СН3-С=О +АТФ
СООН СООН СООН
В результате превращения глюкозы в пировиноградную кислоту клетка накапливает и хранит энергию в виде молекул АТФ.[51].
 Кроме углеродных соединений таутомерия свойственна многим другим классам органических веществ. Как правило, легко диссоциируют протоны, связанные с атомами О, S, Р, N.
Кроме углеродных соединений таутомерия свойственна многим другим классам органических веществ. Как правило, легко диссоциируют протоны, связанные с атомами О, S, Р, N.
Достаточно полно изучены таутомерные превращения азотсодержащих органических соединений. Амиды кислот претерпевают амидо-имидную перегруппировку:
RC=О-NН2 D RC=NН-ОН
Обычно амидные формы устойчивее имидных на » 20 ккал/моль. Для бензоил-a-аминоэтилбензола удалось выделить в чистом виде обе формы:
С6 Н5-С=О(NН)-СН(СН3)-С6Н5 с т. п.128° С и С6Н5-С(ОН)=N-СН(СН3)-С6Н5 с т. пл.123° С. В бензольных растворах преобладает амидная форма, в спиртовых - имидная.[52].
Пиримидины и пурины, входящие как основания в состав нуклеиновых кислот, способны образовывать множество таутомеров. Часто наблюдается лактим-лактамная таутомерия в молекуле урацила:
![]()
![]()
![]()
![]() СН= СН CH = CH
СН= СН CH = CH
![]()
![]()
![]() НО-С N D O=C N-H
НО-С N D O=C N-H
![]() N - C—ОН NH - C=O
N - C—ОН NH - C=O
Она зависит от температуры, природы растворителя, от рН среды, от степени связывания с белками. Например, в нейтральной среде преобладает лактамная форма урацила. Азотистое основание аденин образует водородные связи с цитозином, а его таутомер-иминоформа может образовывать комплементарную пару только с тимином:
![]() NH2 NH
NH2 NH
![]()
![]()
![]()
![]()
![]() C C
C C
![]()
![]()
![]()
![]()
![]() N C - N = CH DH-N C - N =CН
N C - N = CH DH-N C - N =CН
![]()
![]()
![]()
![]() НC C - N Н C C -N
НC C - N Н C C -N
N H N Н
В ряде случаев это может привести к мутациям. Таутомерные превращения характерны и для производных никотинамида, играющих важную роль в окислительных процессах, происходящих в клетке. Примером является система оксипиридин-пиридон [53]. Исследованием ИК-спектров установлено, что равновесие сдвинуто в сторону лактамной формы, т. е. пиридона. Во многих растворителях между молекулами этой формы возникают межмолекулярные водородные связи. [54].
ТАУТОМЕРИЯ ФОС И ВОДОРОДНЫЕ СВЯЗИ
Таутомерии ФОС посвящены многочисленные исследования и с сотрудниками. В основу их положена идея, что кето-енльное равновесие подчиняется закономерностям теории кислотно-основного равновесия. Это означает, что в зависимости от природы атома Х (кислород, сера или азот), стоящего при атоме трёхвалентного фосфора, кислотные свойства веществ могут сильно меняться:
 ( RO)2 Р =Х D( RO)2 Р- ХН
( RO)2 Р =Х D( RO)2 Р- ХН
I H II
Структура диалкилфосфористых кислот изучалась многими физико-химическими методами: парахорами, ИК-спектрами, спектрами комбинационного рассеяния света, с помощью ЯМР-спектроскопии. Показано, что равновесие смещено в сторону «кето-формы», т. е. четырёх координированного фосфора (I), имеющего меньшую кислотность. ЯМР-спектроскопия, применённая к исследованию диалкилфосфористых кислот ( хим. сдвиг –7- 10 м. д.), показывает отсуствие трёхзамещённых фосфитов ( хим. сдвиг -80 –200 м. д.). Это доказывает, что фосфористая, диалкилфосфористая, алкилфосфонистая кислоты находятся более чем на 95% в форме I. Некоторые исследователи считают, что диалкилфосфиты ассоциированы в кольчатые структуры с водородными связями [55]:
![]()
![]() (RО)2Р=О...Н-Р-(ОR)2
(RО)2Р=О...Н-Р-(ОR)2
Н… О
С другой стороны они присоединяют серу, лучше в основных растворителях (диоксан, триэтиламин), однохлористую медь, образуют соли с алкоголятами и сильными основаниями, что с определённостью говорит о смещении равновесия и о наличие формы II. Диалкилфосфористые кислоты легко вступают реакции с альдегидами и кетонами.[56]. Одни легко без катализаторов, другие в присуствии щелочных катализаторов. С хлорзамещёнными альдегидами и кетонами реакция проходит без катализаторов. С хлоралем реакции протекают с большим выделением тепла и с хорошим выходом образуются эфиры a-окси-b,b,b-трихлорэтилфосфиновой кислоты. Некоторые исследователи считают, что эти реакции проходят с енольной формой кислоты II. Нуклеофильный атом трёхвалентного фосфора взаимодействует с электрофильным атомом углерода карбонильной группы с образованием эфиров a-оксиалкилфосфиновых кислот по схеме:
![]() Н
Н
![]()
![]()
![]() (RO)2 Р=Х D(RO)2 Р-ХН + RCН=Оž ( RO)2 Р+-ХН š (RO)2Р-СН-R
(RO)2 Р=Х D(RO)2 Р-ХН + RCН=Оž ( RO)2 Р+-ХН š (RO)2Р-СН-R
СНR-О-- Х… Н О
Ещё легче вступают в реакцию с фосфитами диацетил, ацетилацетон, бензальальдегид, циклогексанон. Изучение специфики водородных связей показало, что a-оксиалкилфосфиновые кислоты способны образовывать как внутримолекулярные, так и межмолекулярные водородные связи. Показано, что протоноакцепторная способность фосфорильной группы оказывает влияние на самоассоциацию a - фосфорилированных спиртов.
Другой пример таутомерии - это фосфорилированные уксусные альдегиды и имины. Показано, что возрастание основности фосфорильного кислорода приводит к увеличению энергии водородных связей, стабилизирующих енольные формы. Фосфорилированные альдегиды стабилизируются преимущественно путём межмолекулярной ассоциации, но с увеличением температуры равновесие смещается в направлении цис-енольной формы с внутримолекулярной водородной связью:
![]()
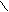
![]()
![]() HC=CHOH… H CH= CH
HC=CHOH… H CH= CH
R2 P=O D R2 P(O)CH2C=O D R2 P=O…H-O
Аналогично имины фосфорилированных уксусных альдегидов проявляют имино-енаминную таутомерию [57]:
![]()
![]()
![]()
![]() R2PCH-CH=NR DR2P - CR= CH
R2PCH-CH=NR DR2P - CR= CH
O R О.. .Н-N-R
![]()
![]() Те же реакции протекают с диалкил - и диарилтиофосфористыми кислотами. Эфиры фосфористой, тиофосфористой и фосфинистых кислот способны к таутомерным превращениям и вступают в реакции с олефинами, ацетиленами, с кетенами, хинонами, диазометаном, карбодиимидами, кетокарбоновыми кислотами и т. д. как в енольной-форме, так и в кетонной. Описано присоединение ди( b-хлорэтил) фосфористой кислоты к ацетилену в присуствии перекисей бензоила и кумила.[58]. Гомолитические реакции протекают по следующей схеме: R· + НРR2 ® RН + · РR2
Те же реакции протекают с диалкил - и диарилтиофосфористыми кислотами. Эфиры фосфористой, тиофосфористой и фосфинистых кислот способны к таутомерным превращениям и вступают в реакции с олефинами, ацетиленами, с кетенами, хинонами, диазометаном, карбодиимидами, кетокарбоновыми кислотами и т. д. как в енольной-форме, так и в кетонной. Описано присоединение ди( b-хлорэтил) фосфористой кислоты к ацетилену в присуствии перекисей бензоила и кумила.[58]. Гомолитические реакции протекают по следующей схеме: R· + НРR2 ® RН + · РR2
![]() О О
О О
НСºСН + ·Р=R2®Н2С=СН-Р=R2
![]()
![]() О О
О О
Относительные скорости реакций зависят от отрыва водорода от ДАФ, при этом в качестве радикалов-катализаторов были использованы Рh·, t-ВuO· , RCОО·, RS· и др. Фотолиз растворов ДАФ в присуствии ловушки t-BuNO приводит к продуктам присоединения - стабильным фосфонильным радикалам нитроксидного типа, фиксируемых методом ЭПР:
t-BuNO + Р·=Z R2 ®t-Bu-N( O·) - P=ZR2
Спектры ЭПР нитроксильных радикалов с атомом фосфора Р непосредственно у радикального центра –N(O·)-Z характеризуются величинами аN, равными 9,9-14,7 э, и аР не более 13,8 э [ 59]. Эти данные говорят о слабом переносе спиновой поляризации на атом фосфора.
Производные фосфора легко вступают в гомолитические реакции, если у атома фосфора находится не только атом водорода, но и атом галогена (Br, CI). Возможно, что реакции окислительного хлорфосфорилирования, заключающиеся во взаимодействии углеводородов с трёххлористым фосфором и кислородом [60], протекают также по радикальному механизму: РСI3 ® РСI2· + CI· и RH + РСI2· + О2 ®RРСI2 , PCI3 + изопрен, а также некоторые другие реакции:
![]()
![]() RОРСI2® RРСI2, или R2С=N-O-РR2® R2C=N-РR2,
RОРСI2® RРСI2, или R2С=N-O-РR2® R2C=N-РR2,
О О
![]()
![]() или ( RO)3Р hn® (RO)2РR, (RO)3 Р + ССI4®(RO)2 РСI,
или ( RO)3Р hn® (RO)2РR, (RO)3 Р + ССI4®(RO)2 РСI,
О О
(RO)3Р + RSSR®(RO)2 Р=S SR [61] и т. д.
В отличие от диалкилтиофосфитов основной формой диалкиланилидофосфитов является форма II с трёхвалентным фосфором.
![]() (RО)2Р=NС6Н5D(RO)Р-NН-С6Н5
(RО)2Р=NС6Н5D(RO)Р-NН-С6Н5
Н I II
Изучение химических свойств последних показало, что им присущи все реакции соединений трёхвалентного фосфора: они присоединяют серу, реагируют с фенилазидом, вступают в арбузовские перегруппировки.[62].
Вода и водородные связи играют значительную роль при установлении таутомерного равновесмя. и сотрудниками изучено таутомерное равновесие диалкилтиофосфатов и алкилалкилтиофосфинатов потенциометрическим методом:

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 |
