

Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
(RO)2Р=S(ОН)D(RO)2Р=О( SH) и R(OR)Р=S(OH)DR(OR)Р=O(SH)
I II I II
При этом использовалось уравнение Гаммета и константа s, специально выведенная для тиокислот фосфора. Исследования проводились в 7% и 80%-ном водном спирте и показали преимущественное наличие тионной формы (I). В водных растворах увеличивается содержание тиольной формы. Дальнейшие исследования с использованием фотометрического метода и основных красителей в бензоле и хлорбензоле показали, что равновесие монотиокислот фосфора практически полностью смещено в сторону тионной формы
( RО)2Р=S-О - · Аr3C+ · Н2О. Очевидно, это связано с большей основностью Р=О связи по сравнению с Р=S связью, которая подтверждается тем, что фенол образует водородную связь с Р=О ( энергия ~3 ккал/моль), а Р= S нет.
Изучение реакций солей этих кислот показало, что они ведут себя как амбидентные анионы, т. е. способны реагировать по обоим концам триады в зависимости от жёсткости реагента[63]:
R2Р=ОS-D R2Р=SО - + RХ→ R2Р=О(SR)
R2 Р=ОS-DR2Р=SO - + R3 O+ → RP=O(SR) и RP=S(OR)
R2 P=OS-DR2P=SO - + RCOCI→ RP=S(OC=OR)
ВОДА И ВОДОРОДНЫЕ СВЯЗИ В ДЕЙСТВИИ
Земная кора образовалась и начала охлаждаться около 4 млрд. лет назад. Образование океанов и накопление первичной атмосферы Земли произошло около 3,5 млрд. лет назад. В этой атмосфере, очевидно с малым количеством кислорода, началось образование исходных органических молекул таких, как формальдегид, цианистый водород и их производных. Благодаря воде и водородным связям эти молекулы превратились в аминокислоты, сахара, нуклеиновые кислоты, коацерваты, в комплексы. Коацерваты, выделившись из окружающего раствора, могли взаимодействовать и с окружающей средой. Так в процессе эволюции возникли первичные живые организмы, обладающие определённым метаболизмом.
На сегодня существует несколько научных теорий о происхождении жизни. Современной наукой доказано, что основой жизни на Земле являются микроорганизмы. Миллиарды их населяют почву, воду. Они приспособились к очень жёстким условиям: могут сохраняться в космическом холоде, в отсутствии кислорода, без света. Известны микроорганизмы, живущие в вулканах при температуре ~200°С и превращающие сероводород в сульфаты. Открыты микроорганизмы в глубинных шахтах, которые превращают водород и углерод в метан, т. е. неорганические вещества в органику. Известны рачки, существующие в сверхсолёных водоёмах. А сколько организмов существуют в океанах на глубинах свыше 250 м при низких температурах и в отсутствии света! Наверное - миллиарды. Вслед за микроорганизмами такое же распространение на Земле получили грибы. И все они имели одного предка - живую клетку.
На основе этих фактов предложены две гипотезы возникновения жизни на Земле. По первой, она была занесена метеоритными дождями с других планет Солнечной системы, что подтверждается последними исследованиями комет. В Мерчисонском метеорите, который упал в Австралии в 1969 г., найдены аминокислоты, в других пурины, пиримидины, углеводороды. Обнаружены метеориты с остатками организованной материи - бактериями и водорослями.
По другой гипотезе, высказанной , на Земле появились приемлемые условия для возникновения живой клетки: это ультрафиолетовые лучи, тепло, углекислый газ, азот, вода, водород, метан. То, что из неорганических веществ можно получить органические вещества, стало известно с начала XIX века. В 1824 году немецкий химик Ф. Вёлер синтезировал гидролизом неорганического дициана органическое вещество - щавелевую кислоту, а несколько позже из циановокислого аммония получил мочевину - продукт жизнедеятельности животного организма. Это был удар по распространённому виталистическому учению о «жизненной силе». В то время многие химики утверждали, что получить мочевину без помощи почек и вообще без участия какого-либо живого существа не возможно. В 1862 г. химик Бертело, пропуская водород между угольными электродами, осуществил синтез органического вещества ацетилена. Впоследствии при прохождении электрического тока между угольными электродами в атмосфере азота и водорода была получена синильная кислота.
2С + N2 + Н2® 2 НСN.
В 1953 г. американский учёный С. Миллер поместил смесь газов, состоящую из водорода, метана и аммиака, в закрытый сосуд с водой и пропускал через неё электрический разряд в течение 20 часов. Проанализировав продукты реакции, Миллер обнаружил в растворе более 8 различных аминокислот, которые, как известно, представляют собой основной строительный материал живой клетки. После этого стало понятно, что для образования аминокислот и живой клетки не требуется никаких идеалистических сил, кроме обычных физических и химических, существующих на Земле (взрывов, молний, УФ-лучи и т. д.). Из-за отсуствия в атмосфере озонового слоя миллиарды лет назад УФ-лучи, очевидно, обладали более высокой энергией, чем сегодня. Это способствовало превращению простых молекул в более сложные соединения через радикальные реакции. Наиболее важными молекулами для строительства клетки являлись углеводороды, формальдегид и цианистый водород.[64]. Реакции могли происходить по следующим схемам:
СН4 ®hv ·CН3 + ·Н СН4 + ·СН3 ®СН3-·СН2 + Н2
2 Н2О®hv 2НО· + 2Н·
·СН3 + НО·® СН3ОН ® hv ·CН2ОН ·С2Н5 + НО·®С2Н5ОН ®hv C2Н4·ОН
НО-СН2· + НО·®НО-СН2ОН® Н2С=О ·С2Н4ОН + НО·® СН3-СН=О
Цианистый водород образуется под действием УФ-излучения из метана и аммиака в несколько стадий. СН4 + NH3®hv Н-СºN + 6Н·
Синтез альдегидов и цианистого водорода дало ключ к появлению основных составляющих жизни - белков, нуклеиновых кислот, ферментов. Одна из схем получения аминокислот из альдегидов, аммиака, цианистого водорода и воды включает следующие реакции :
2R-CН=О + NН3® R-CН= NНR-CH=NH + HCN®
R-CH-NH2CN + H2O® R-CH-NH2-CONH2+ H20 ® R-CHNH2-COOН+NH3
Высокая полярность нитрильной группы, её лёгкая поляризуемость под действием различных реагентов предопределяет возможность вступления циановодорода в реакции присоединения. Вторая стадия – нуклеофильная атака углеродного атома имина ионом цианида может происходить как с фронта, так и с тыла, что приводит к образованию L и D стереоизомеров. При синтезе аминокислот в живом организме эта реакция происходит на поверхности ассиметрического катализатора - фермента, поэтому образуется только один энантиомер
(L). Аминонитрилы превращались в аминокислоты в результате гидролиза. Последний может протекать в нейтральной, кислой и щелочной средах. Вначале образуется иминоспирт, который под действием второй молекулы воды гидролизуется в аминокислоту с выделением аммиака. Реакцию ускоряют кислоты:
![]()
![]()
![]() RCH-(NH2)CºN+НОН ® RCH(NH2)C=NH+НОН D RCH-CNH2 +HOHš
RCH-(NH2)CºN+НОН ® RCH(NH2)C=NH+НОН D RCH-CNH2 +HOHš
NH2 О
® RCHNH2COOH + NH3.
Аминокислоты существуют преимущественно в виде биполярных ионов (цвиттерионов). Полимеризация ста и более аминокислот в пептидную цепь образует первичную структуру белка. В этой схеме аминонитрилы могли полимеризоваться ещё до реакции гидролиза: RCH(NH2)CºN+H2NRCHCºN® RCH(NH2)C=NHNНСНR-CºN + Н2О ®
®RCH(NH2 )C=ONHCHR-CºN и т.д.
Вода и водородные связи принимают активное участие в образовании нуклеиновых кислот. Строительными кирпичиками молекул нуклеиновых кислот служат нуклеотиды. Нуклеотид состоит из трёх компонентов - пуринового или пиримидинового основания, сахара-рибозы и фосфорной кислоты. Соединяясь между собой через рибозу, эти три компонента могут полимеризоваться через фосфат и сахар, образуя огромные молекулы с боковыми цепями со структурой пуринов и пиримидинов. Так, молекула аденина могла возникнуть из пяти молекул циановодорода, а рибоза - из пяти молекул формальдегида:
![]()
![]()
![]() основание основание основание
основание основание основание
-фосфат-сахар-фосфат-сахар-фосфат-сахар-
5 H-CºN® Аденин
5 СН2=О®Рибоза
Сахара, имеющие пять или шесть атомов углерода, защищаясь от дальнейших нуклеофильных атак, оказались склонны к циклизации в ацетальные формы. Такова схематичная картина образования аминокислот, нуклеотидов и живой клетки.
Вода сыграла значительную роль и в метаболизме первых живых организмов на Земле. Они появились в отсуствии кислорода и поэтому должны были получать энергию за счёт анаэробного разложения молекул пищи. Этот древнейший механизм жизни состоял в расщеплении молекул глюкозы на две молекулы пировиноградной кислоты с помощью ферментов. Выделившаяся энергия запасалась в виде АТФ. Но так как её не хватало, то организмы разработали процесс фотосинтеза, в ходе которого использовалась энергия солнца и углекислый газ. Первая стадия фотосинтеза состояла в поглощении солнечной энергии с помощью хлорофилла. Последний представляет комплексное соединение иона магния и порфирина. Фотовозбуждённый хлорофилл поставлял энергию, необходимую для превращеня АДФ в АТФ:
АДФ + НОР=О(ОН )2hv® АТФ
В зелёных растениях при фотосинтезе происходит восстановление СО2 до формальальдегида, при этом вода служит источником водорода, а в атмосферу выделяется кислород:
НОН + О=С=Оhv® H2C=O + O2
Этот процесс достаточно сложный, протекает через ферментативный катализ, его можно изобразить схематично:
СО2+дифосфатрибулозы® фосфоглицериновая кислота®фосфоглицериновый альдегид®углеводороды.
Процесс фотосинтеза привёл не только к созданию нового источника пищи, но и сделал доступным выделяющийся при фотосинтезе кислород для других форм жизни.[65].
Вода явилась основой развития многообразных форм жизни на земле как флоры, так и фауны, чему безусловно способствовали постоянно изменяющиеся условия жизни -вулканическая деятельность, потепления и похоладания климата, приспособляемость биологической жизни к любым изменениям среды на протяжении миллионов лет. Об этом свидетельствует теория Ч. Дарвина, представленная в его книге « Происхождение видов», над которой он работал более 20 лет.
Вода, являясь прекрасной средой для обитания микроорганизмов, была ответственна во время развития пандемий. Чума -«чёрная смерть» унесла во времена средневековья сотни миллионов человек. Человечество тяжело страдало от грязной воды, несущей такие болезни, как дизентерия, холера, брюшной тиф, малярия, грипп. Открытие сульфаниламидов и антибиотиков, бурное развитие химии лекарственных средств, чистая вода сокрушили эти инфекции.
Сегоднейшие успехи биохимии - открытие носителей наслественной информации - биологических полимеров ДНК и РНК, синтез гена человека, развитие новой науки - генной инженерии должно обязательно привести к резкому увеличению продолжительности жизни человека. С другой стороны, вода, как и атмосферный азот, в будущем может стать основой энергетики Земли, так как при её химическом распаде образуется энергетически важный водород, что подтверждается следующими химическими реакциями:
2 Н2О ®щёлочь, электролиз 2Н2 + О2 или С + 2Н2О1000°® 4Н2 + СО2 или
СН4 + Н2О1000° Ni® 3Н2 +СО N2 +4H2O +4V(OH)2Mg↔H2NNH2(NH3) + 4V(OH)3
ГИДРОЛИЗ. РЕАКЦИИ ГИДРАТАЦИИ
Гидролиз галоидных алкилов в водных растворах щелочей может протекать как по механизму SN1, так и по механизму SN2. Здесь многое зависит от строения алкильного радикала. Первичные алкилы гидролизуются по механизму SN2:
CH3 Гаl + HO-® HO-….СН3….Гаl-® HO-CH3 + Гаl -
Третичные- по механизму SN1:
R3C-Гаl ® Гаl - …С+R3®-ОН НО-СR3 .
По механизму SN1 начальная ионизация алкилгалогенида осуществляется за счёт энергии сольватации образующихся ионов, и поэтому существенно зависит от полярности и диэлектрической проницаемости растворителя. Так, при переходе от этанола к 50% водному этанолу скорость гидролиза трет-бутилхлорида, протекающего по механизму SN1,возрастает в 30000 раз. Катион, атом углерода которого несёт положительный заряд, представляет собой карбониевый ион, при этом атом углерода иона переходит из тетраэдрического состояния в более устойчивое - планарное. Так как атака нуклеофила может происходить и с фронта и с тыла, то в случае оптического карбаниона получаются рацематы.
В случае механизма SN2 происходит обращение конфигурации атома углерода. Известно большое число реакций замещения, но некоторые из них имеют одну особенность. Если рядом с реакционным центром расположены атомы, обладающие собственной электронной парой О, S, N, то они способны действовать в качестве собственного нуклеофила. Так, при щелочном гидролизе b-хлоргидрина
![]()
![]() ( CICH2CH2OH→CICH2CH2O-→CH2 –CH2→HOCH2CH2OH )
( CICH2CH2OH→CICH2CH2O-→CH2 –CH2→HOCH2CH2OH )
O
первая стадия состоит в превращении его в алкоксид-ион. Этот ион атакует атом углерода, несущий хлор, с тыла по механизму SN2, что приводит к обращению конфигурации и к образованию окиси этилена. Трёхчленное кольцо вновь атакуется гидроксил-ионом, причём атакуется менее замещённый атом углерода по механизму SN2 и вновь с обращением конфигурации. Аналогичный механизм гидролиза характерен и для соединений, содержащих в галоидгидринах вместо кислорода атомы серы или азота. Правильность рассмотренного механизма доказывается тем, что выделены трёхчленные циклы в виде эпоксидов, этиленсульфидов, этиленимидов.
Участие в реакциях соседних групп может резко изменять скорость процесса. В одних и тех же условиях скорость гидролиза СICH2CH2SC2H5 происхоит в 10000 раз быстрее, чем его кислородного аналога. Такое невозможно объяснить ни индуктивным, ни стерическими эффектами. Гидролиз, возможно, связан с образованием промежуточного продукта – сульфониевого иона циклического строения. Такое образованное кольцо, будучи сильно напряжённым, легко подвергается гидролизу:
![]()
![]() EtS-CH2CH2-CI®Et - S+-CH2
EtS-CH2CH2-CI®Et - S+-CH2
CH2…CI-НОН®EtSCH2CH2-OH + Н+
Иприт в малых концентрациях (доли мг) практически гидролизуется водой мгновенно, но если иприт находится в каплях или при его массвых проливах, гидролиз иприта провести достаточно сложно из-за его гидрофобности. Реакции гидролиза идут на поверхности и не до конца, поэтому необходимо использовать перемешивание, эмульгаторы, щёлочь, моноэтаноламин и т. д. Соответствующие азотсодержащие соединения также прояляют склонность легко гидролизоваться через соли этилениммониевого типа:
![]() R2N-CH2CH2CI®R2N+- CH2…СI - HOH®R2NCH2CH2-OH
R2N-CH2CH2CI®R2N+- CH2…СI - HOH®R2NCH2CH2-OH
![]() CH2
CH2
Но в случае аминов гидролиз протекает медленее из-за более высокой устойчивости промежуточного циклического соединения. В отличие от серы атом кислорода в аналогичных хлоргидринах не склонен отдавать свою электронную пару углероду и не образует циклический ион, поэтому замещение хлора на гидроксильную группу в этих соединениях протекает гораздо труднее.[66].
ГИДРАТАЦИЯ ДВОЙНОЙ и ТРОЙНОЙ СВЯЗЕЙ
Алкены легко превращаются в спирты в результате либо непосредственного присоединения воды, либо присоединения серной кислоты как катализатора с последующим гидролизом.
RСН=СН2 + Н2О +H ®RСНОНСН3
Присоединение воды к алкинам особенно легко протекает в присутствии солей двухвалентной ртути. Эта реакция открыта М. Кучеровым в 1880 г. Присоединение воды протекает по правилу Марковникова. При этом сначала образуется енол (ненасыщенный спирт), который быстро изомеризуется в карбонильное соединение. Из ацетилена получается уксусный альдегид, из алкилацетиленов – кетоны, а из винилацетилена в промышленности получают метилвинилкетон.[149].
НСºСН + Н2О® HgSO4 [ CH2=CHOH ]nCH3C=OH
![]() CH3-CºCH+H2O®HgSO4[CH3-C=CH2]nCH3C=OCH3
CH3-CºCH+H2O®HgSO4[CH3-C=CH2]nCH3C=OCH3
OH
Вода легко писоединяется и к карбонильным соединениям. Карбонильная группа имеет плоское строение. Атом углерода в ней находится в sp2 –гибридном состоянии. Он образует три σ- связи, оси которых расположены в одной плоскости под углом 120º друг к другу. Негибридизированная р-орбиталь атома углерода, занятая одним электроном, и р-орбиталь кислорода, содержащая один неспаренный электрон, в результате бокового перекрывания образуют π- связь. В отличие от двойной связи связь С=О полярна. Электронная плотность в карбонильной группе смещена к атому кислорода, так как атом кислорода являетсяболее электроотрицательным, чем атом углерода. Последнее приводит к появлению частичного положительного заряда на атоме углерода и отрицательного заряда на атоме кислорода. Пониженная электроння плотность на атоме углерода объясняет склонность этих соединений присоединять нуклеофильные агенты, воду.
Формальдегид в воде почти целиком существует в виде гидрата.
СН2О + Н2ОD Н2С(ОН)2
Ацетальдегид в воде гидратируется только на ~ 50%, а ацетон не гидратируется совсем. Образующиеся гидратные формы обычно неустойчивы, выделить их не удаётся. Наличие электроноакцепторных заместителей в алкильных группах облегчает гидратацию и стабилизирует образующие гидраты. Так, глиоксаль, хлораль, индантрион-1,2,3 (нингидрин) образуют кристаллические гидраты, котрорые могут быть выделены:
НС=О-НС=О +НОНšНС=О-СН(ОН)2
СCI3 -НC=O + НОНšССI3 –CH(OH)2
C6 H4(C=O)3 + НОН š С6 Н4 (С=О)2 С(ОН)2
Эти гидраты, вероятно, стабилизируются за счёт образования водородных связей между гидроксильными группами и атомами кислорода или хлора соседних a-углеродных атомов.
Реакции присоединения НСN, НSO3- могут служить типичными примерами реакций SN 2 присоединения анионов:
Н+
R-C=O-HHCNn[R-CH-O - CN]®R-CHOHCN.
Реакции альдегидов со спиртами протекают сложнее, чем с водой. Вначале образуются полуацетали, причём на скорость их образования очень сильно влияют структурные ( стерические ) факторы как со стороны спирта, так и со стороны альдегида (кетона ). Циклический полуацеталь из хлораля и этиленгликоля был выделен в кристалическом состоянии.
НОСН2-СН2ОН + О=СНССI3šНОСН2СН2ОСН(ОН)ССI3
Cкорости образования полуацеталей резко возрастают в присуствии незначительных количеств кислот. Низкие значения изменения энтропии при образовании полуацеталей и ацеталей дали возможность предположить механизм с циклическим переносом электронов.
=С=О + R-OH š =C(ОН)ОR
С другой стороны, реакции образования и гидролиза ацеталей на первый взгляд казавшимися простыми, являются сложными и трудно объяснимыми. Многочисленными исследованиями показано, что они идут с разрывом карбонильной связи по смешанному SN1 и SN2 механизму. Показано, что первая стадия образования карбониевого иона мономолекулярна и определяет скорость реакции гидролиза, а вторая бимолекулярна. Это подтверждено реакцией ацеталя с оптически активным спиртом D-(+)-2-октанолом. Оказалось, что при этом образуется спирт с той же величиной вращения, что и исходный. Изучение гидролиза ацеталей в воде, обогащённой 18О, дало спирты, не содержащие меченого кислорода.
Н+
![]() R2C(OR)2 + H318O+D[R2C(OR)2 ]’R2C-OR HОН šR2C=O18 + 2ROH
R2C(OR)2 + H318O+D[R2C(OR)2 ]’R2C-OR HОН šR2C=O18 + 2ROH
НО - ОН
Внутримолекулярные взаимодействия влияют на гидролиз ацеталей. Скорость гидролиза метилтиоуксусного ацеталя на два порядка выше, чем метоксиуксусного, что можно объяснить образованием циклического иона на стадии, определяющей скорость реакции [67]:
![]()
![]() CH3SCH2CH(ОR)2+Н+(ОН-)®CH3S+- CH2®CH3SCH2CH=O
CH3SCH2CH(ОR)2+Н+(ОН-)®CH3S+- CH2®CH3SCH2CH=O
CHOR
Гидролиз сложных эфиров кислот может протекать как в присуствии кислоты, так и в щелочной среде (омыление). Реакции обычно включают разрыв ацильной связи:
 RC=O-OR + HOH +H+DRC=O-ORD RC-ORD RC=O-OH + ROH
RC=O-OR + HOH +H+DRC=O-ORD RC-ORD RC=O-OH + ROH
Н+ ОН
![]()
![]() RC=O18-OR + НOH’R-C-OR’RC-O-+ R18OH
RC=O18-OR + НOH’R-C-OR’RC-O-+ R18OH
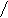
![]() 18О ОН О
18О ОН О
Таким образом, при действии нуклеофильных агентов на соединения RC=O-X реакция начинается с атаки на положительно заряженный карбонильный атом. Это взаимодействие в большинстве случаев определяет скорость процесса. Атака происходит почти под прямым углом к плоскости, в которой лежит молекула RCOХ и сопровождается возникновением связи между углеродом карбонильной группы и нуклеофилом с одновременным отходом Х в виде аниона. Переходное состояние характеризуется sp3- гибридизацией электронных орбит углерода, связи которого направлены к вершинам тетраэдра. Таким образом, структура переходного состояния предусматривает раскрытие карбонильной связи ( С=О ) и образование промежуточного соединения с четырёхкоординированным атомом углерода. Группы X в соединениях RC=OX, благодаря индуктивному эффекту и эффекту сопряжения, увеличивают или уменьшают этот заряд. Чем больше +М и +I-эффекты групп или атомов, связанных с карбонилом, тем меньше его реакционная способность. По убыванию последней основные классы веществ, содержащих карбонильную группу, можно расположить в ряд: -С=О-CI > С=О-СN > - HC=O-> RC=O>-C=O-OR. Наибольшую активность проявляет карбонильная группа хлорангидридов кислот, так как хлор имеет сильный –I –эффект, наименее активны анионы органических кислот. Скорость реакций карбонильных соединений зависит также от стерических факторов, нуклеофильности агента и сольватационных эффектов.
Дегидратация.
Внутримолекулярная дегидратация спиртов осуществляется в присуствии катализаторов при нагревании и приводит к образованию алкенов. Отщепление воды от нессиметричных спиртов протекает по правилу Зайцева: водород отщепляется от наименее гидрогенизированного атома углерода с образованием наиболее разветвлённого алкена. Дегидратацию обычно проводят, нагревая спирт с серной или фосфорной кислотой, либо пропуская пары спирта над оксидом алюминия (АI2O3) при 350-400°C. Наиболее легко реакции дегдратации протекают у третичных спиртов, несколько труднее - у вторичных и значительно труднее - у первичных спиртов. При дегидратации спиртов используют также кислые катализаторы на основе Al2O3, ДМSO, фталевый ангидрид, ионообменные смолы типа дауэкс 50 и т. д. [146].
СН3СН(ОН)СН2СН3®катализ. t. СН3СН=СНСН3
Межмолекулярная дегидратация спиртов или кислот происходит при использовании тех же катализаторов, но при более низких температурах и приводит к образованию простых и сложных эфиров, либо ангидридов кислот, например:

![]()
![]() СН2СН2СООН СН2СН2С=О
СН2СН2СООН СН2СН2С=О
![]() СН2СН2СООН®130° - НОН О
СН2СН2СООН®130° - НОН О
СН2СН2С=О
ГИДРОЛИЗ И НЕКОТОРЫЕ ПЕРЕГРУППИРОВКИ
ТРЁХВАЛЕНТНЫХ ФОС
Химия фосфорорганических соединений (ФОС ) представляет в настоящее время обширную область элементорганической химии, весьма интересную в теоретическом и практическом отношении. Систематическое изучение ФОС было начато более 150 лет назад работами Михаэлиса. Известно широкое применение ФОС в различных областях медицины, сельского хозяйства, промышленности. Основу ФОС составляют вещества с трёх - и четырёх - координированным атомом фосфора. Реакции гидролиза трёхвалентных ФОС из-за их не стабильности обычно сопровождаются перегруппировкой в производные пятивалентного фосфора. Они напрямую связаны с широко известной перегруппировкой в её известном классическом виде[68]. При этом малоустойчивая конфигурация трёхвалентного фосфора 3s23р3 переходит через переходное состояние в sp3-гибридизацию тетракоординированного атома фосфора:
![]()
![]() (RO)2 PCI + H2 O® (RO)2 P+ OH CI - ® (RO)2P=O + HCI
(RO)2 PCI + H2 O® (RO)2 P+ OH CI - ® (RO)2P=O + HCI
H H
Действие воды на полные эфиры фосфористой кислоты начал изучать ещё в 1905 г. . Он показал, что (СН3О)3Р практически мгновенно и нацело превращается в диметилфосфит. Гомологи метилового эфира, а именно этиловый эфир и другие полные эфиры трёхвалентного фосфора оказались стойкими веществами, вода не оказывала на них действие даже при нагревании до 100 °С. Зато реакция в присуствии кислот протекала быстро и до конца:
(RO)3 P + H2 O+HCI®HPO(OR)2 + ROH +HCI
Амидоэфиры фосфористой кислоты также легко подвергаются гидролизу, алкоголизу и вступают в Арбузовскую перегруппировку[69].
![]()
![]()
![]() (RO)2PNR2 + H2O®(RO)2 P - NR2 ®(RO)2-P=O +HNR2
(RO)2PNR2 + H2O®(RO)2 P - NR2 ®(RO)2-P=O +HNR2
H OH H
Алкоголиз некоторых амидов трёхвалентного фосфора пропаргиловым спиртом проводился нами при °С. С количественным выходом фракционной перегонкой были выделены соединения не трёхвалентного фосфора, а b-диалкиламино-b-метилвинил-фосфонаты. Очевидно, образующиеся на первой стадии пропаргилфосфиты с высокой скоростью изомеризуются в алленилфосфонаты, а выделяющиеся вторичные амины, не успев покинуть зону реакции, присоединяются к непредельной связи:
![]()
![]()
![]() (RO)2PNR2+HOCH2CºCH - HNRR®(RO)2POCH2CºCH®HC=C=CHP(OR)2 ® +HNRR ® CH3C=CНP(OR)2 O
(RO)2PNR2+HOCH2CºCH - HNRR®(RO)2POCH2CºCH®HC=C=CHP(OR)2 ® +HNRR ® CH3C=CНP(OR)2 O
NRR O
Температурные условия алкоголиза зависят как от строения амидофосфитов, так и от строения ацетиленового спирта. Накопление амидных групп при атоме фосфора облегчает течение реакции. Взаимодействие трис-(диметиламидо)фосфита с пропаргиловым спиртом протекает при комнатной температуре, а с третичным ацетиленовым спиртом при нагревании до 150° в ампуле.[70]: (CH3)2N)3P + HOCR2CºCH®(CH3)2N)2POCR2CºCH®
(R2 N)2P+… -O
![]() - CH=C=CR2НNRR ®(R2N)2PCH=C(NR2)-CHR2
- CH=C=CR2НNRR ®(R2N)2PCH=C(NR2)-CHR2
O
Изучение этих реакций с помощью ИК-спектроскопии во времени в термостатированной кювете в толуольном растворе при 30° по частотам nºСH 3300cм-1, nС=С=С 1970 cм-1 и n С=С-N 1570 cм-1 показало, что присоединение вторичных аминов происходит по алленовому, а не по изомерному ему a, b-пропинильному радикалу, т. е. в данных примерах переход алленовой структуры в a, b-пропинильный радикал не происходит. Строение фосфорилированных енаминов было доказано гидролизом их в b-кетофосфонаты, так как они легко оттитровывались водной кислотой :
![]()
![]() (RO)2P-CH=C(CH3)NR2®HCI (RO)2P-CH=C(CH3)NR2HCI НОН®( RO)2PCH2CCH3
(RO)2P-CH=C(CH3)NR2®HCI (RO)2P-CH=C(CH3)NR2HCI НОН®( RO)2PCH2CCH3
![]()
![]() O O O O
O O O O
Ацетилен - алленовая перегруппировка эфиров Р3 протекает очень энергично, иногда со взрывами. А. Пудовику удалось выделить несколько пропаргиловых эфиров трёхвалентного фосфора, но лишь в сыром виде.[75]. Б. Ионину с сотрудниками удалось синтезировать и выделить дихлорпропаргилфосфит, который при стоянии в течение 10 дней изомеризовался в дихлорангидрид алленилфосфоновой кислоты [75]:
НСºС-СН2ОН +РСI3 пиридин®HCºC-CH2OPCI2®H2C=C=CHP=OCI2
В плане исследования механизма термической перегруппировки пропаргиловых эфиров кислот трёхвалентного фосфора нами было изучено взаимодействие D2-пропаргилового спирта с трёххлористым фосфором. Спирт был приготовлен 6-кратной обработкой тяжёлой водой пропаргилового спирта. Реакцию проводили при минус 30°С в присуствии триэтиламина. В результате реакции был выделен дихлорангидрид a-дейтероалленилфосфоновой кислоты:
DCºCCH2OD + PCI3 ® ( DCºCCH2OPCI2 ) ® H2C=C=CDP=OCI2
I II III
Изомеризацию II не удалось осуществить в эфире при длительном нагревании, а после удаления эфира она проходила со взрывом. Изомеризация проведена при нагревании II в бензоле при 60-70°С в течение 12 часов. [71]. Мы считали, что на изомеризацию определённое влияние оказывает ацетиленовый протон, но оказалось, что в реакцию вовлекаются и g - замещённые пропаргиловые спирты. Так, продукты взаимодействия диалкилхлорфосфитов с 4-N, N-дилкиламинобутин-2-олом-1 также претерпевают термическую изомеризацию с образованием О, О-диалкил-a(N, N-диалкиламинометил)алленилфосфонатов по схеме [72] :
(RO)2PCI + HOCH2CºCCH2NR2 ®(RO)2POCH2CºCCH2NR2 ®
CH2=C=CP(OR)2
![]()
![]() R2N-CH2 O
R2N-CH2 O
Попытки замедлить изомеризацию пропаргилфосфитов в производные четырёхкоординационного фосфора введением электроотрицательных заместителей к атому фосфора привела к неоднозначным результатам. Оказалось, что пропаргиловые эфиры кислот трёхвалентного фофора, содержащие трифторметильные радикалы, также легко изомеризуются в производные четырёхкоординационного фосфора.[73].
СF3PI2+2HOCH2CºCH+2N(C2H5)2®CF3P(OCH2CºCH)2®
![]() CF3P=O(OCH2CºCH) + 2HIN(C2H5)3
CF3P=O(OCH2CºCH) + 2HIN(C2H5)3
CH=C=CH2
Аналогично был выделен О-этилтрифторметилалленилфосфинат. Реакция бис(трифторметил)иодфосфина с пропаргиловым спиртом привела к бис-(трифторметил)-a,b-пропинилфосфиноксиду:
(CF3)2PI + HOCH2CºCH + N(C2H5)3 ®(CF3)2P(О)-CºC-CH3 + HINR3
Пропаргиловый эфир трёхвалентного фосфора I удалось выделить в виде пирокатехинового производного по схеме [74]:
![]()
![]() C6H4O-PCI+HOСH2CºCH® С6Н4-О-P-OCH2CºCH.
C6H4O-PCI+HOСH2CºCH® С6Н4-О-P-OCH2CºCH.
![]()
![]() O O I
O O I
Очистка продукта I осуществлялась перекристаллизацией из эфира при низких температурах. С помощью ИК-спектроскопии изучена кинетика перегруппировки его в пирокатехиновые эфиры алленил - и пропинилфосфоновых кислот:
![]()
![]() =P-OCH2CºCH® = P-CH=C=CH2→=P-CºCCH3
=P-OCH2CºCH® = P-CH=C=CH2→=P-CºCCH3
I O O
Ацетилен-алленовая перегруппировка - это реакция первого порядка, что свидетельствует о внутримолекулярном механизме, высказанном ранее [75]. Нам не удалось наблюдать ацетилен – алленовую изомеризацию на производных трёхвалентного фосфора, так как она вторична и происходит одновременно с переходом трёхвалентного фосфора в пятивалентный. При этом промежуточный комплекс содержит повидимому не первичную ацетиленовую структуру, как показано в литературе[75 ], а уже алленовую, которая менее жёсткая, нежели ацетиленовая, и может легче подойти к трёхвалентному атому фосфора:
![]()
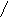 (RO)2P-OCR2C≡CH→ (RO)2-P+-O- ® (RO)P-C=C=CR2
(RO)2P-OCR2C≡CH→ (RO)2-P+-O- ® (RO)P-C=C=CR2

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 |
