

Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
МЕТОДИЧЕСКИЕ УКАЗАНИЯ ПО ВЫПОЛНЕНИЮ КОНТРОЛЬНЫХ ЗАДАНИЙ
, Рябцева : Методические указания для решения задач.
ОГЛАВЛЕНИЕ
Введение
Список использованных обозначений
1. Основные классы неорганических соединений
1.1. Оксиды
1.2. Гидроксиды
1.3. Кислоты
1.4. Соли
2. Взаимодействие веществ
2.1. Химическая термодинамика
2.1.1. Основные понятия
2.1.2. Энергетика химических процессов (термохимические расчеты). Первый закон термодинамики
2.1.3. Энтропия. Химическое сродство. Второй закон термодинамики
2.1.4. Условия самопроизвольного протекания процессов. Третий закон термодинамики
2.1.5. Примеры решения задач
2.2. Химическая кинетика
2.2.1. Закон действия масс (закон Гульдберга-Вааге)
2.2.2. Влияние температуры на скорость реакции
2.2.3. Химическое равновесие. Константа равновесия
2.2.4. Смещение химического равновесия. Принцип Ле Шателье
2.2.5. Примеры решения задач
3. Растворы и реакции в водных растворах
3.1. Концентрации растворов
3.1.1. Способы задания концентрации растворов
3.1.2. Закон эквивалентов
3.1.3. Пример решения задачи
3.2. Теория растворов
3.2.1. Давление пара растворов
3.2.2. Кипение и замерзание растворов
3.2.3. Осмос. Осмотическое давление
3.2.4. Количественные характеристики растворов электролитов. Закон растворения
3.2.5. Произведение растворимости. Условие образования осадка
3.2.6. Примеры решения задач
3.3. Ионное произведение воды и водородный показатель. Гидролиз солей
3.3.1. Ионное произведение воды и водородный показатель
3.3.2. Гидролиз солей
3.3.3. Примеры решения задач
3.4. Комплексные соединения
3.4.1. Общие понятия о структуре комплексного соединения
3.4.2. Номенклатура комплексных соединений
3.4.3. Диссоциация комплексных соединений в водных растворах
3.4.4. Разрушение комплексных ионов
3.4.5. Примеры решения задач
3.5. Жёсткость природных вод
3.5.1. Общие понятия о жесткости воды
3.5.2. Способы умягчения воды
3.5.3. Примеры решения задач
4. Окислительно-восстановительные системы
4.1. Окислительно-восстановительные реакции
4.1.1. Общие понятия об окислительно-восстановительных реакциях
4.1.2. Подбор коэффициентов методом электронного баланса
4.1.3. Подбор коэффициентов методом электронно-ионного баланса
4.1.4. Пример решения задачи
4.2. Гальванические элементы и электрохимические процессы
4.2.1. Понятие электрода
4.2.2. Типы электродов. Электродный потенциал
4.2.3. Гальванические элементы
4.2.4. Электролиз расплавов и водных растворов электролитов
4.2.5. Законы Фарадея
4.2.6. Примеры решения задач
Приложения
Литература
СПИСОК ИСПОЛЬЗОВАННЫХ ОБОЗНАЧЕНИЙ
А - механическая работа;
G - изобарно-изотермический потенциал (свободная энергия Гиббса);
Ж - жёсткость воды;
γ – температурный коэффициент скорости химической реакции;
С – концентрация раствора;
СВ - молярная концентрация (молярность) раствора;
См – моляльная концентрация (моляльность раствора);
СЭ (допускается СН) – молярная концентрация эквивалентов вещества (эквивалентная концентрация, нормальная концентрация, нормальность раствора);
Н - энтальпия;
h – степень гидролиза раствора соли;
i – изотонический коэффициент раствора;
КД – константа диссоциации вещества в растворе электролита;
КР – константа равновесия;
КН - константа нестойкости комплексного иона;
k – константа скорости химической реакции;
kЭ – эбуллиоскопическая постоянная вещества растворителя;
kК – криоскопическая постоянная вещества растворителя;
МВ - молярная масса вещества;
Мэкв – молярная масса эквивалентов вещества;
mA - масса растворителя в растворе;
mB - масса растворённого в растворе вещества;
mP - масса раствора;
Р – давление;
pH – водородный показатель;
Q - теплота;
ρ - плотность раствора;
S - энтропия;
U - внутренняя энергия;
V - объем;
υ – скорость химической реакции;
ω – массовая доля растворённого вещества.
1. ОСНОВНЫЕ КЛАССЫ НЕОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
1.1. Оксиды
Оксиды – соединения элементов с кислородом с общей формулой ![]() .
.
Элементы могут быть металлами и неметаллами. Например: СаО – оксид кальция; N2O – оксид азота (I); N2O5 – оксид азота (V). Кислород в оксидах двухвалентен. Если для элемента характерна переменная валентность, то она обозначена в скобках.
Оксиды не диссоциируют на ионы, т. к. не являются электролитами.
Оксиды подразделяются на:
- солеобразующие:
· основные,
· кислотные,
· амфотерные;
- несолеобразующие.
Основными оксидами называют те, которые с водой образуют основания (образованы металлами в низших степенях окисления) (+1; +2; редко +3):
Li2O + H2O ![]() 2LiOH
2LiOH
оксид лития гидроксид лития
Кислотными оксидами называют оксиды, которые при взаимодействии с водой образуют кислоты или соответствуют кислотам.
Кислотные оксиды образуются неметаллами:
N2O5 соответствует азотная кислота HNO3
оксид азота (V)
или металлами в высших степенях окисления (+3; +4; +5; +6; +7):
Mn2O7 соответствует марганцовая кислота HMnO4
оксид марганца (VII)
Амфотерные оксиды соответствуют некоторым металлам (Pb, Sn, Al, Zn...): PbO – оксид свинца (II); ZnO – оксид цинка.
Амфотерным оксидам соответствуют гидроксиды, обладающие основными и кислотными свойствами:
PbO → Pb(OH)2 – обладает основными и
кислотными свойствами
Несолеобразующими оксидами называют оксиды, которые не образуют и не соответствуют ни кислотам, ни гидроксидам:
СО N2O NO
оксид углерода (II) оксид азота (I) оксид азота (II)
1.2. Гидроксиды
Гидроксиды – вещества, отвечающие составу ![]() .
.
Гидроксиды – электролиты, которые в воде диссоциируют на ионы металлов и гидроксид-ионы (![]() ).
).
NaOH – гидроксид натрия;
Fe(OH)3 – гидроксид железа (III).
Именно гидроксид-ионы обуславливают общие свойства класса гидроксидов, включая окраску индикаторов.
Гидроксиды подразделяются на основные, кислородсодержащие кислоты и амфотерные.
Основные гидроксиды соответствуют основным оксидам и называются основаниями:
K2O – KOH; MgO – Mg(OH)2
Гидроксиды-основания могут быть хорошо и плохо растворимыми в воде.
Хорошо растворимые в воде гидроксиды называются щелочами.
Щелочи образуются металлами, располагающимися в главных подгруппах первой и некоторых металлов второй групп периодической таблицы и их восемь: LiOH; NaOH; KOH; RbOH; CsOH; FrOH; Ba(OH)2 и Ca(OH)2.
Щелочи являются сильными электролитами, диссоциирующими в водных растворах необратимо:
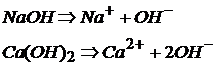
Все остальные основания являются трудно растворимыми в воде соединениями и слабыми электролитами:
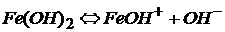
ион гидроксожелеза(II)
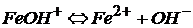
Существуют два исключения:
NH4OH – гидроксид аммония – хорошо растворимое в воде соединение, но слабый электролит (![]() – ион аммония);
– ион аммония);
Ca(OH)2 – трудно растворимое в воде соединение, но сильный электролит.
Кислородосодержащие кислоты соответствуют кислотным оксидам:
![]()
![]() Cr(OH)6 C(OH)4
Cr(OH)6 C(OH)4
CrO3 – или CO2 – или
![]()
![]() H2CrO4 ·2H2O H2CO3 · H2O
H2CrO4 ·2H2O H2CO3 · H2O
Диссоциация кислот описана ниже в разделе 1.3.
Амфотерные гидроксиды соответствуют амфотерным оксидам и обладают свойствами как оснований так и кислородосодержащих кислот:
![]() Sn(OH)2 гидроксид олова (II) – основание
Sn(OH)2 гидроксид олова (II) – основание
SnO –
![]() H2SnO2 – оловянистая кислота
H2SnO2 – оловянистая кислота
Как основание гидроксид Sn(II) диссоциирует:
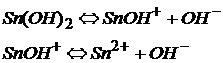
Как кислота гидроксид Sn(II) диссоциирует:
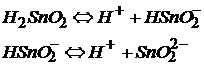
1.3. Кислоты
Кислота – сложное вещество, в молекуле которого имеется один или несколько атомов водорода, которые могут быть замещены атомами (ионами) металлов или аммонием.
Оставшаяся часть молекулы кислоты называется кислотным остатком.
По количеству частиц водорода, способных замещаться в химических реакциях, кислоты подразделяются на одно-, двух-, и многоосновные:
HNO3; HCl; HCN – одноосновные кислоты;
H2SO4; H2CO3; H2S – двухосновные кислоты.
Как электролиты кислоты могут быть сильными и слабыми.
Кислоты – сильные электролиты:
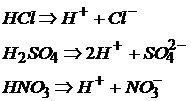
Кислоты – слабые электролиты диссоциируют ступенчато:
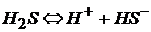
гидросульфид ион
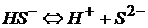
сульфид ион
По природе кислотного остатка кислоты подразделяются на бескислородные:
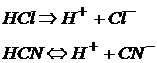
и кислородсодержащие:
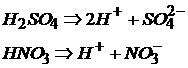
Названия кислот определяются природой кислотного остатка:
H2S – сероводородная кислота;
HCN – циановодородная кислота.
Названия кислородсодержащих кислот зависят от степени окисления центрального атома кислотного остатка.
Если центральный атом находится в высшей степени окисления, то название кислоты имеет окончание «ная»:
HNO3 – азотная.
Если центральный атом кислотного остатка находится не в наивысшей степени окисления, то название кислоты имеет окончание «истая»:
HNO2 – азотистая.
1.4. Соли
Все соли – сильные электролиты, образующиеся в результате реакции нейтрализации кислоты основанием.
Соли подразделяются на:
· средние,
· кислые,
· основные,
· двойные,
· смешанные.
Средние соли образуются, если гидроксид и кислота взаимодействуют в эквивалентном отношении. Молекула средней соли состоит из катиона металла или аммония и кислотного остатка кислоты:
MgCO3 – карбонат магния;
(NH4)2S – сульфид аммония.
Диссоциируют средние соли на ионы металла (аммония) и кислотного остатка.
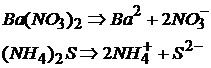
Кислые соли возможны для многоосновных кислот (H2SO4; H3PO4 …), в которых ионы Н+ частично замещены на ионы металла или аммония:
NaHCO3 – гидрокарбонат натрия.
В названии кислой соли присутствует приставка «гидро».
NH4H2PO4 – дигидрофосфат аммония.
Кислые соли диссоциируют на ионы металла (аммония) и кислотный остаток, включающий одну или несколько частиц водорода:
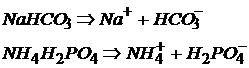
Основные соли образуются в том случае, если гидроксид является многокислотным (Fe(OH)2, Al(ОН), а кислота взята в недостатке:
Al(ОН)3 + НСl Al(ОН)2 Сl + Н2О или, что то же самое:
Аl ОН ОН ОН + НCl Al(ОН)2 Сl + Н2О.
В названии основной соли присутствует приставка «гидроксо». Присутствие нескольких ![]() ионов обозначается греческим числительным.
ионов обозначается греческим числительным.
Al(OH)2Cl – хлорид дигидроксо алюминия;
(Fe(OH)2)2SO4 – сульфат дигидроксо железа (III).
Основные соли диссоциируют на сложные катионы, состоящие из иона металла и гидроксогруппы и ионы кислотного остатка:
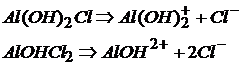
Соли, образованные двумя металлами и одной кислотой, называются двойными солями.
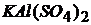 – сульфат кальция алюминия (алюмокальциевые квасцы):
– сульфат кальция алюминия (алюмокальциевые квасцы):
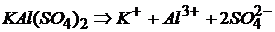 .
.
Соли, образованные одним катионом и двумя разными кислотами, называются смешанными.
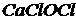 (или
(или 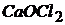 ) – кальциевая соль хлороводородной (
) – кальциевая соль хлороводородной (![]() ) и хлорноватистой (
) и хлорноватистой (![]() ) кислот:
) кислот:
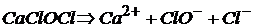 .
.
2. ВЗАИМОДЕЙСТВИЕ ВЕЩЕСТВ
2.1. Химическая термодинамика
Химическая термодинамика изучает превращение энергии при химических реакциях и способность химических систем совершать полезную работу.
2.1.1. Основные понятия
Термодинамические параметры системы – это величины, которые характеризуют состояние системы и могут быть непосредственно измерены. Основными параметрами состояния являются: давление Р, объем V, концентрация С.
Термодинамические функции системы – это переменные величины, которые зависят от параметров состояния. К термодинамическим функциям относятся: внутренняя энергия U, теплота Q, механическая работа А, энтальпия Н, энтропия S, изобарно-изотермический потенциал (свободная энергия Гиббса) G и др. Термодинамические функции бывают двух видов: функции состояния и функции процесса.
К функциям состояния относятся такие, изменения которых не зависят от пути и способа проведения процесса, а зависят только от начального и конечного состояния системы. Так, например, если функция состояния внутренняя энергия в начальном состоянии системы равна U1, а в конечном U2, то изменение ∆U = U2 – U1 не зависит от того как осуществляется процесс.
В противоположность функциям состояния изменение функций процесса зависит от того, при каких условиях и каким путем протекал процесс. К функциям процесса относится теплота Q и механическая работа А.
Состояние системы – это совокупность ее физических и химических свойств.
Термодинамический процесс – это изменение системы, связанное с изменением хотя бы одного термодинамического параметра или функции. В зависимости от способа проведения процессы могут быть термодинамически обратимыми или необратимыми.
2.1.2. Энергетика химических процессов (термохимические расчеты). Первый закон термодинамики
При любом процессе соблюдается закон сохранения материи и выполняется первое начало термодинамики, как частный случай этого закона:
Теплота, поглощаемая системой, расходуется на увеличение внутренней энергии и на совершаемую системой механическую работу.
Первый закон термодинамики применим к процессам, сопровождающимся преобразованием теплоты.
Q = ∆U + А,
где А – работа выполненная системой против действия внешних сил.
Если такая работа сводится к расширению или к сжатию, то такой силой является внешнее постоянное давление
Q = ∆U + Р∆V,
где ∆V – изменение объема системы.
Применение первого начала термодинамики к различным процессам. При изохорном процессе ∆V = 0. Уравнение первого начала термодинамики имеет вид:
Qν = ΔU
Поскольку внутренняя энергия является функцией состояния, для изохорных процессов количество теплоты также не зависит от пути перехода и определяется только начальным и конечным состоянием системы. Из уравнения следует, что в изохорном процессе вся поглощаемая теплота расходуется на увеличение внутренней энергии системы.
Прежде чем рассматривать применение первого закона термодинамики к изобарному процессу, познакомимся с широко применяемой термодинамической функцией, называемой энтальпией Н. Она определяется как сумма полной внутренней энергии системы и произведение внешнего давления на объем, который занимает система:
Н = U + РV
Для макропроцессов:
Qp = ΔH,
где Qp – количество поглощенной или выделенной системой теплоты при постоянном внешнем давлении.
Энтальпия является функцией состояния системы. Значит, в изобарном процессе количество поглощенной или выделенной теплоты не зависит от способа проведения процесса и определяется только начальным и конечным состоянием системы. Видно, что в изобарном процессе (∆Р = 0) количество теплоты измеряется изменением энтальпии.
При изотермическом процессе внутренняя энергия – величина постоянная, ∆U = 0. Тогда уравнение первого начала:
QT = А
где QT – количество поглощенной или выделенной системой теплоты при постоянной температуре.
Следовательно, при изотермическом процессе поглощенная теплота расходуется только на совершаемую системой механическую работу.
При адиабатном процессе, когда Q = 0, уравнение принимает вид:
А = ─∆U
Это означает, что в адиабатном процессе механическая работа может совершаться только за счет внутренней энергии системы.
Термохимические расчеты основаны на законе Гесса:
Тепловой эффект химической реакции не зависит от пути, по которому она протекает, а определяется только природой, состоянием и количествами исходных веществ и продуктов реакции.
Для термохимических расчетов чаще используют следствие из закона Гесса:
Тепловой эффект химической реакции равен сумме стандартных теплот образования продуктов реакции, за минусом суммы теплот образования исходных веществ, с учетом стехиометрических коэффициентов уравнения реакции
Изменение энтальпии в реакции ![]() :
:
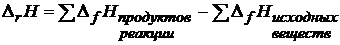
Здесь теплотой образования ![]() (индекс f – от английского «formation» – «образование») данного сложного вещества называется тепловой эффект реакции образования 1 моль этого вещества из простых веществ. Например:
(индекс f – от английского «formation» – «образование») данного сложного вещества называется тепловой эффект реакции образования 1 моль этого вещества из простых веществ. Например:
С + О2 = СО2; 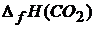 =
=![]()
Теплота образования простых веществ приравнена в термодинамике к условному нулю.
В справочных таблицах приводятся стандартные теплоты образования многочисленных сложных веществ 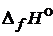 . Стандартные условия T = 298 К и P = 101,3 кПа (1 атм).
. Стандартные условия T = 298 К и P = 101,3 кПа (1 атм).
Правило знаков: тепловой эффект эндотермической реакции, т. е. реакции происходящей с поглощением теплоты, будет положительным (![]() > 0), а тепловой эффект экзотермической реакции, протекающей с выделением теплоты – отрицательным (
> 0), а тепловой эффект экзотермической реакции, протекающей с выделением теплоты – отрицательным (![]() < 0).
< 0).
2.1.3. Энтропия. Химическое сродство. Второй закон термодинамики
Самопроизвольно могут протекать реакции, сопровождающиеся не только выделением, но и поглощением теплоты. Тенденцию системы к беспорядку характеризует величина, которую называют энтропией. Энтропия S, так же как внутренняя энергия U, энтальпия Н, объем V и др., является свойством вещества, пропорциональным его количеству и обладающим аддитивностью (суммируется при соприкосновении систем). Энтропия отражает движение частиц вещества и является мерой неупорядоченности системы. Она возрастает с увеличением движения частиц: при нагревании, испарении, плавлении, расширении газа и т. п. Процессы, связанные с упорядоченностью системы: конденсация, кристаллизация, полимеризация, сжатие – ведут к уменьшению энтропии. Энтропия является функцией состояния, т. е. ее изменение (∆S) зависит только от начального и конечного состояния системы и не зависит от пути процесса.
Изменение энтропии в ходе химической реакции может быть вычислено по следствию из закона Гесса
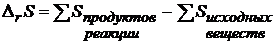
Энтропия 1 моль вещества в его стандартном состоянии при соответствующей температуре называется стандартной энтропией ![]() . Стандартные энтропии многих простых и сложных веществ приводятся в таблицах. Следует подчеркнуть, что речь идет об абсолютных энтропиях, а не их изменении, сопровождающем образование сложного вещества из простых веществ (вспомним теплоты образования). Поэтому стандартные энтропии простых веществ не равны нулю.
. Стандартные энтропии многих простых и сложных веществ приводятся в таблицах. Следует подчеркнуть, что речь идет об абсолютных энтропиях, а не их изменении, сопровождающем образование сложного вещества из простых веществ (вспомним теплоты образования). Поэтому стандартные энтропии простых веществ не равны нулю.
Второе закон термодинамики определяет возможность самопроизвольного протекания процессов, удовлетворяющих условию:
![]() >
> ![]() ,
,
где ![]() – тепловая энергия, которой обменивается система с окружающей средой при постоянной температуре.
– тепловая энергия, которой обменивается система с окружающей средой при постоянной температуре.
При суммировании абсолютных энтропий каждая из них должна быть умножена на соответствующий стехиометрический коэффициент.
2.1.4. Условия самопроизвольного протекания процессов. Третий закон термодинамики
В изолированных системах самопроизвольно протекают процессы, сопровождающиеся увеличением энтропии (ΔS ≥ 0). Это следует из второго закона термодинамики. Когда энтропия достигает максимального для данных условий значения, в системе установится состояние равновесия (ΔS = 0). Однако, известны многочисленные процессы, происходящие самопроизвольно, несмотря на уменьшение энтропии (ΔS < 0), например кристаллизация жидких веществ. Возможность таких процессов определяет третий закон термодинамики.
При постоянных давлении и температуре самопроизвольно могут протекать только процессы, сопровождающиеся уменьшением G.
Это формулировка третьего начала термодинамики.
Пределом протекания таких процессов, то есть условием равновесия является минимальное значение энергии Гиббса. То есть выполнение условия:
ΔG = 0.
Термодинамическая функция G – изобарно-изотермический потенциал (свободная энергия Гиббса):
G = H – T ∙ S.
Изменение энергии Гиббса для макропроцессов определяется формулой Гиббса:
∆G = ∆H – T ∙∆ S.
Изменение энергии Гиббса реакции можно также рассчитать на основании 3-го следствия из закона Гесса:
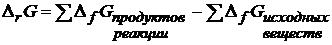
с учетом стехиометрических коэффициентов реакции.
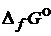 (вещества) – стандартная энергия Гиббса образования вещества (приведены в таблицах).
(вещества) – стандартная энергия Гиббса образования вещества (приведены в таблицах).
Таким образом, условием самопроизвольного протекания изобарно-изотермических процессов является не положительность изменения энергии Гиббса:
∆G < 0.
Если ∆G < 0, процесс принципиально осуществим; если ∆G > 0 процесс самопроизвольно протекать не может. Чем меньше ∆G, тем сильнее стремление к протеканию данного процесса и тем дальше он от состояния равновесия, при котором ∆G = 0 и ∆H = T ∙ ∆S. Из соотношения ∆G = ∆H – T ∙∆S видно, что самопроизвольно могут протекать и процессы, для которых ∆H > 0 (эндотермические). Это возможно, когда ∆S > 0, но ![]() >
> ![]() , и тогда ∆G < 0. С другой стороны, эндотермические реакции (∆H < 0) самопроизвольно не протекают, если при ∆ S < 0 окажется, что ∆G > 0.
, и тогда ∆G < 0. С другой стороны, эндотермические реакции (∆H < 0) самопроизвольно не протекают, если при ∆ S < 0 окажется, что ∆G > 0.
Для процессов с одинаковыми знаками ∆H и ∆S существует температура, называемая критической:
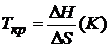 ,
,
определяющая состояние равновесия системы (∆H = T ∙ ∆S).
При температурах, больших критической возможны эндотермические процессы, сопровождающиеся увеличением энтропии (∆ S > 0).
При температурах, меньших критической возможны экзотермические реакции, сопровождающиеся уменьшением энтропии (∆ S < 0).
2.1.5. Примеры решения задач
Задача 1. Рассчитать тепловой эффект реакции
(н. у.),
используя значения теплот образования веществ – участников реакции.
Решение. Согласно следствию из закона Гесса
![]() =
= 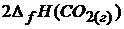 +
+ 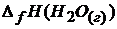 –
– 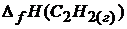 –
–
Табличные значения теплот образования всех веществ – участников реакции
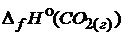 = -393,5
= -393,5 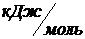 ;
; 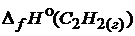 = -226,8
= -226,8 ![]() ;
;
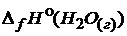 = -241,8 ;
= -241,8 ; 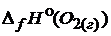 = 0.
= 0.
Подставляя значения стандартных теплот образования в уравнение, получаем:
![]() = 2 ∙ (-393,5) + (-241,8) – 226,8 – 2,5 ∙ 0 = -1295 кДж
= 2 ∙ (-393,5) + (-241,8) – 226,8 – 2,5 ∙ 0 = -1295 кДж
Ответ: тепловой эффект реакции равен ![]() = -1295 кДж.
= -1295 кДж.
Задача 2. Определить количество теплоты, выделившейся при сжигании 5 литров этана при нормальных условиях, если в ходе реакции образуются пары воды и углекислый газ.
Решение.
Чтобы найти тепловой эффект реакции, определим изменение энтальпии реакции, используя следствие из закона Гесса и справочные значения стандартных теплот образования соответствующих веществ.
= -393,5 ; 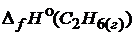 = -84,7
= -84,7 ![]() ;
;

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 10 11 |
