

Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
величина ![]() задается выражением
задается выражением
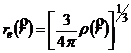 . (15)
. (15)
В (14) обменные эффекты заключены в ![]() , а все корреляционные эффекты определяются множителем
, а все корреляционные эффекты определяются множителем ![]() .
.
В общем случае довольно сложно записать выражение для Exc, однако в [7] показано, что для систем с медленно меняющейся плотностью обменно-корреляционная энергия может быть представлена в виде:
![]() , (16)
, (16)
где ![]() – обменно-корреляционная энергия, приходящаяся на один электрон, однородного газа плотности
– обменно-корреляционная энергия, приходящаяся на один электрон, однородного газа плотности ![]() .
.
Из стационарности (12) следует, что
![]() . (17)
. (17)
Тогда можно записать [7]:
![]() , (18)
, (18)
здесь ![]() , а
, а ![]() – вклад обмена и корреляции в химический потенциал однородного газа плотности
– вклад обмена и корреляции в химический потенциал однородного газа плотности ![]() .
.
Далее для заданных ![]() и
и ![]() можно найти плотность
можно найти плотность ![]() и полную энергию
и полную энергию ![]() , решая самосогласованно одночастичное уравнение Шредингера:
, решая самосогласованно одночастичное уравнение Шредингера:
![]() , (19) где
, (19) где 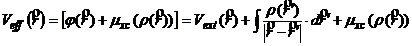 , (20)
, (20)
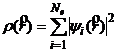 , (21)
, (21)
Ne – число электронов. Множители ![]() образуют спектр энергий одночастичных состояний. Таким образом, теория функционала плотности сводит многоэлектронную задачу к одноэлектронной с эффективным локальным потенциалом
образуют спектр энергий одночастичных состояний. Таким образом, теория функционала плотности сводит многоэлектронную задачу к одноэлектронной с эффективным локальным потенциалом 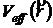 .
.
Из (19) следует [8]:
![]()
![]() . (22)
. (22)
Тогда полная энергия для системы запишется в следующем виде [6]:
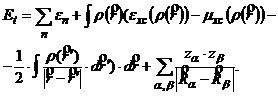 (23)
(23)
Последнее слагаемое является результатом вклада взаимодействия остова атома a (ему соответствует радиус-вектор ![]() ) c остовом атома b (ему соответствует радиус-вектор
) c остовом атома b (ему соответствует радиус-вектор ![]() ). В (23) подразумевается суммирование по спиновым переменным.
). В (23) подразумевается суммирование по спиновым переменным.
Выбор базисных функций.
Выбором базисных атомных функций в приближении линейной комбинации атомных орбиталей (ЛКАО) определяется, насколько точно ряд ЛКАО аппроксимирует молекулярную орбиталь (МО). Этот ряд должен достаточно быстро сходиться, т. е. малое число атомных орбиталей (АО) должно аппроксимировать МО с требуемой точностью. Существует три основных критерия для выбора базисных функций:
1. Базисные функции должны давать в основном хорошее приближение к истинной волновой функции.
2. Базисные функции должны допускать аналитическое вычисление нужных интегралов.
3. Полное число базисных функций не должно быть очень большим.
Любую непрерывную функцию можно разложить по полному набору функций. Например, непрерывную функцию одной переменной ![]() можно разложить в ряд по полиномам Лягерра:
можно разложить в ряд по полиномам Лягерра:
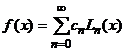 . (24)
. (24)
Функцию нескольких переменных также можно разложить по каждому ее аргументу в такой же ряд:
![]() . (25)
. (25)
Можно использовать такие ряды в разложении молекулярной орбитали. Трудность состоит в том, что не для всякой функции эти ряды быстро сходятся, т. е. небольшое количество членов хорошо аппроксимирует функцию ![]() . Для разложения волновой функции в такой ряд необходимо брать большое число членов (1000 и более), чтобы получить достаточно хорошие результаты.
. Для разложения волновой функции в такой ряд необходимо брать большое число членов (1000 и более), чтобы получить достаточно хорошие результаты.
Наиболее быстро сходящиеся ряды строятся на базисе атомных орбиталей. Лучшими атомными функциями являются самосогласованные атомные орбитали, вычисленные Э. Клементи и Р. Ватсоном путем решения уравнений Хартри – Фока для атомов. Однако эти функции получаются не в аналитическом виде, а в табличном. Проводить расчеты с функциями, заданными в числовом виде, крайне трудно и неудобно. В качестве базиса АО можно использовать слэтеровские орбитали. Однако слэтеровская АО плохо описывает хартри – фоковскую АО вблизи ядра. В то же время две слэтеровские АО достаточно хорошо аппроксимируют точную хартри – фоковскую функцию атома, в связи с чем был предложен весьма распространенный дубль-зета-базис (DZ), где каждая атомная функция аппроксимируется двумя слэтеровскими функциями с разными экспонентами.
Расчеты можно разделить на два класса: расчеты с минимальным атомным базисом и с расширенным. Минимальный базисный ряд состоит только из АО внутренних и валентных оболочек свободных атомов, расширенный базис включает дополнительно атомные орбитали, не занятые в основном состоянии. Расчеты с минимальным базисом легче, однако расшиненный базис дает более точные результаты. Основная сложность заключается в вычислении кулоновских и обменных интегралов, вычисления которых на слэтеровских функциях чрезвычайно сложны и трудоемки. Для упрощения расчетов Бойс использовал набор гауссовского типа для аппроксимации каждой слэтеровской АО:
![]() , (26)
, (26)
где 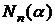 – нормировочный множитель;
– нормировочный множитель; ![]() – варьируемый параметр.
– варьируемый параметр.
Преимущество такой замены заключается в том, что вычисление интеграла становится значительно проще. Основной недостаток гауссовских функций в том, что они плохо отражают поведение АО. Для аппроксимации АО с достаточной точностью необходимо брать большее число гауссовских АО, чем слэтеровских.
Для улучшения описания молекулярных орбиталей на больших расстояниях от ядра часто используют базисные ряды с включением поляризационных функций.
В настоящий момент доступны стандартные базисы на основе слейтеровских (STO) и гауссовых (GTO) орбиталей на сайте http://www. emsl. pnl. gov:2080/forms/basisform. html.
Динамика молекул. Гармоническое приближение.
Ядерная волновая функция ![]() , получаемая из решения уравнения
, получаемая из решения уравнения
![]() , (27)
, (27)
зависит от ![]() переменных
переменных ![]() , из которых только
, из которых только ![]() координат относятся к внутренним координатам, а остальные шесть координат описывают поступательное и вращательное движения молекулы как целой. Оператор
координат относятся к внутренним координатам, а остальные шесть координат описывают поступательное и вращательное движения молекулы как целой. Оператор ![]() можно представить в виде
можно представить в виде
![]() , (28)
, (28)
где 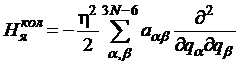
зависит только от внутренних координат ![]() и является оператором кинетической энергии молекулы во внутренних координатах; коэффициенты
и является оператором кинетической энергии молекулы во внутренних координатах; коэффициенты ![]() есть функции от
есть функции от ![]() . Остальные члены
. Остальные члены ![]() ,
, ![]() и
и ![]() относятся к движению молекулы как целого и взаимодействию этого движения с колебаниями. Для малых колебаний влияние вращательного и поступательного движения молекулы на ее колебания мало. Это означает, что последним членом в уравнении (28) можно пренебречь и ядерную функцию можно представить как произведение функций, каждая из которых зависит только от колебательных, поступательных и вращательных координат (степеней свободы), причем только колебательная функция зависит от внутренних координат:
относятся к движению молекулы как целого и взаимодействию этого движения с колебаниями. Для малых колебаний влияние вращательного и поступательного движения молекулы на ее колебания мало. Это означает, что последним членом в уравнении (28) можно пренебречь и ядерную функцию можно представить как произведение функций, каждая из которых зависит только от колебательных, поступательных и вращательных координат (степеней свободы), причем только колебательная функция зависит от внутренних координат:
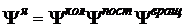 . (29)
. (29)
В этом случае уравнения для каждого из этих движений решаются отдельно и электронная энергия ![]() как функция от внутренних координат входит только в колебательное уравнение:
как функция от внутренних координат входит только в колебательное уравнение:
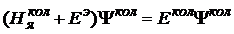 ;
;
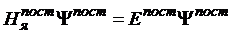 ; (30)
; (30)
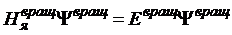 .
.
Полная энергия молекулы ― это сумма отдельных ее компонент:
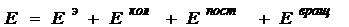 . (31)
. (31)
Уравнение для колебаний молекулы можно записать в виде:
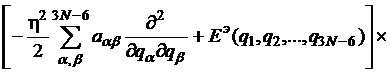
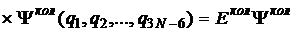 . (32)
. (32)
Можно ввести новые координаты ![]() , которые называют нормальными, тогда (32) распадается на
, которые называют нормальными, тогда (32) распадается на 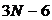 независимых уравнений:
независимых уравнений:
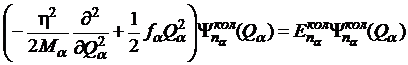 , (33)
, (33)
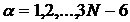 ,
,
которые представляют собой уравнения состояний гармонического осциллятора. Собственные значения ![]() гармонического осциллятора выражаются формулой
гармонического осциллятора выражаются формулой
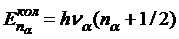 , (34)
, (34)
где ![]() ― частоты нормальных колебаний;
― частоты нормальных колебаний; ![]() ― колебательные квантовые числа, принимающие значения 0, 1, 2, … .
― колебательные квантовые числа, принимающие значения 0, 1, 2, … .
Собственные функции гармонического осциллятора имеют вид
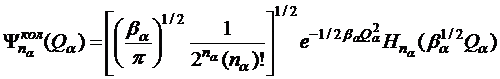 , (35)
, (35)
где 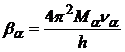 ;
; ![]() ― полином Чебышева – Эрмита степени
― полином Чебышева – Эрмита степени ![]() относительно
относительно ![]() . Формула для полной колебательной энергии молекулы имеет вид
. Формула для полной колебательной энергии молекулы имеет вид
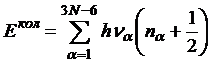 . (36)
. (36)
Каждому колебательному уровню энергии многоатомной молекулы отвечает свой набор колебательных квантовых чисел ![]() отдельных нормальных колебаний. Нижний уровень, для которого все
отдельных нормальных колебаний. Нижний уровень, для которого все 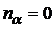 , называют нулевым колебательным уровнем многоатомной молекулы, а энергию, ему соответствующую, ― энергией нулевых колебаний молекулы:
, называют нулевым колебательным уровнем многоатомной молекулы, а энергию, ему соответствующую, ― энергией нулевых колебаний молекулы: 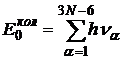 . (37)
. (37)
Таким образом, для того чтобы решить задачу о движении ядер, необходимо рассчитать (в приближении Борна – Оппенгеймера) ППЭ соответствующего электронного состояния в окрестности минимума, аппроксимировать эту ППЭ в окрестности минимума параболоидом (гармоническим потенциалом) и только после этого решать задачу (28) о колебаниях ядер в молекуле. Несмотря на столь многие допущения, к которым приходится прибегать в таких расчетах, вычисленные указанным способом частоты (энергии) колебаний для низших пяти-шести колебательных уровней достаточно хорошо совпадают с экспериментальными значениями.
Практическое занятие. Исследование электронной структуры, динамики молекулярных комплексов N6.
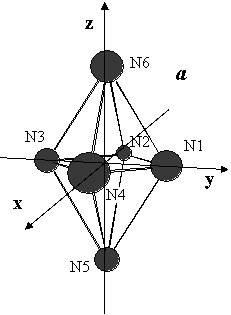
Возможные комплексы ![]() , представлены на рис. 1. В таблице приведены равновесные параметры и полная энергия.
, представлены на рис. 1. В таблице приведены равновесные параметры и полная энергия.
Комплекс | R1, Å | R2, Å | R3, Å | a1, ˚ | a2, ˚ | Etot, a. e. |
| 1.881 | 1.407 | 1.407 | 43.93 | 68.04 | -327.77211 |
| 1.318 | 1.318 | 1.318 | 120.00 | 120.00 | -328.21776 |
| 1.233 | 1.233 | 1.898 | 107.19 | 53.60 | -327.99608 |
| 1.135 | 1.243 | 1.446 | 115.92 | 172.98 | -328.26576 |
| 1.152 | 1.173 | 1.250 | 180.00 | 180.00 | -328.15523 |
| 1.131 | 1.240 | 1.445 | 109.36 | 172.31 | -328.27800 |
| 1.519 | 1.195 | 1.468 | 103.75 | 46.35 | -328.16176 |
| 1.478 | 1.478 | 1.518 | 60.00 | 90.00 | -328.02723 |
| 1.519 | 1.295 | 2.042 | 71.84 | 97.48 | -328.10748 |
| 1.247 | 1.464 | 1.496 | 94.87 | 110.43 | -328.16594 |
| 1.299 | 1.502 | 1.462 | 105.48 | 49.13 | -328.13036 |
| 1.179 | 1.179 | 2.090 | 96.70 | 166.61 | -328.23745 |
| 1.115 | 1.831 | 1.329 | 42.55 | 158.71 | -328.16854 |
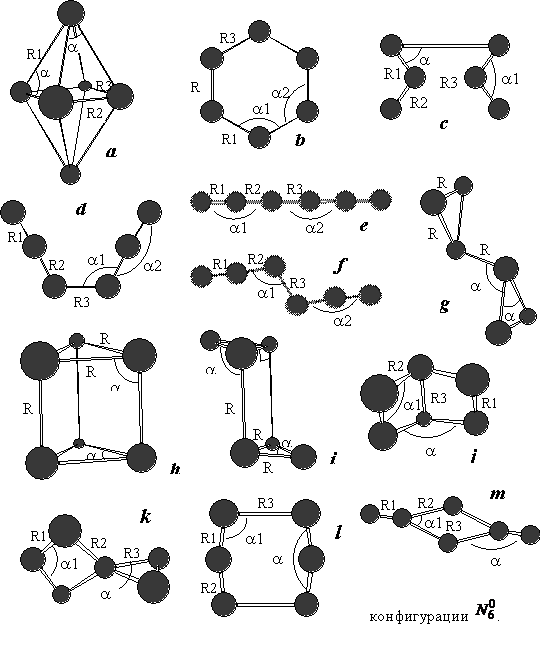 |
Теоретико-групповой анализ молекулярных комплексов N6
1. Структура ![]() (a) имеет группу симметрии
(a) имеет группу симметрии ![]() , которая состоит из элементов:
, которая состоит из элементов: ![]() ,
, ![]() ,
, ![]() ,
, ![]() ,
, ![]() ,
, ![]() ,
, ![]() ,
, ![]() ,
, ![]() ,
, ![]() ,
, ![]() ,
, ![]() ,
, ![]() ,
, ![]() ,
, ![]() ,
, ![]() . В таблице 11 приведены характеры неприводимых представлений.
. В таблице 11 приведены характеры неприводимых представлений.
Характеры неприводимых представлений группы ![]() .
.
|
|
| 2 | 2 |
|
|
| 2 | 2 | |
A1g | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
A1u | 1 | 1 | 1 | 1 | 1 | -1 | -1 | -1 | -1 | -1 |
A2g | 1 | 1 | 1 | -1 | -1 | 1 | 1 | 1 | -1 | -1 |
A2u | 1 | 1 | 1 | -1 | -1 | -1 | -1 | -1 | 1 | 1 |
B1g | 1 | -1 | 1 | -1 | 1 | 1 | -1 | 1 | -1 | 1 |
B1u | 1 | -1 | 1 | -1 | 1 | -1 | 1 | -1 | 1 | -1 |
B2g | 1 | -1 | 1 | 1 | -1 | 1 | -1 | 1 | 1 | -1 |
B2u | 1 | -1 | 1 | 1 | -1 | -1 | 1 | -1 | -1 | 1 |
Eu | 2 | 0 | -2 | 0 | 0 | 2 | 0 | -2 | 0 | 0 |
Eg | 2 | 0 | -2 | 0 | 0 | -2 | 0 | 2 | 0 | 0 |

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 |
