


Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
На правах рукописи
ДАВЫДОВА
Каринэ Сергеевна
СОВЕРШЕНСТВОВАНИЕ СИСТЕМЫ ЭКСПЕРТИЗЫ
ВОСПРОИЗВЕДЕННЫХ ЛЕКАРСТВЕННЫХ СРЕДСТВ ПРИ ИХ
ГОСУДАРСТВЕННОЙ РЕГИСТРАЦИИ
14.03.06 – Фармакология, клиническая фармакология
А в т о р е ф е р а т
диссертации на соискание ученой степени
доктора медицинских наук
Москва-2011
Работа выполнена в Филиале «Клиническая фармакология» Научного
Центра Биомедицинских Технологий РАМН
Научные консультанты:
доктор медицинских наук,
профессор, академик РАМН
доктор фармацевтических наук,
профессор
Официальные оппоненты:
доктор медицинских наук,
профессор, чл-корр. РАМН
доктор медицинских наук,
профессор, чл-корр. РАМН
доктор медицинских наук,
профессор, чл-корр. РАМН
Ведущая организация:
ГОУ ВПО Волгоградский государственный медицинский университет
Защита состоится «__» _________ 2011 г. в ____ часов на заседании Диссертационного 208.04.13 при Первом Московском Государственном Медицинском Университете имени ( Москва, ул. Трубецкая, стр.2).
С диссертацией можно ознакомиться в библиотеке Первого Московского Государственного Медицинского Университета имени Москва, Нахимовский проспект,.
Автореферат разослан «__» ___________ 2011 г.
Ученый секретарь Диссертационного Совета,
доктор медицинских наук Владимир Владимирович Архипов
ВВЕДЕНИЕ
Актуальность темы.
Большая часть лекарственных средств на современном фармацевтическом рынке является воспроизведенными (генерическими) препаратами. Согласно данным розничного аудита (IMS Health и DSM Group), доля генериков в настоящее время составляет от 77 до 88 % в натуральном выражении (, 2008; , , 2009 г.). При этом согласно прогнозам, эта доля будет неуклонно расти. По объему генерического сектора Россия занимает третье место в мире, после Китая и Индии. В то же время, структура рынка стран большой семерки формируется следующим образом: в США — 12% генериков, в Японии — 30%, в Германии — 35%, во Франции — 50%, в Англии — 55%, в Италии — 60%, в Канаде — 64% (, 2008 г).
Основной задачей регуляторных органов в области здравоохранения является обеспечение тщательного контроля качества воспроизведенных лекарственных средств и надлежащей документации, подаваемой в регуляторные органы при регистрации такого лекарственного средства. Генерики, как и инновационные (оригинальные) препараты, должны отвечать общим требованиям, предъявляемым в рамках Общего (или единого) технического документа: эффективность, безопасность, качество.
В настоящее время в России для оценки эквивалентности генерического лекарственного средства и препарата сравнения при их государственной регистрации проводят исследования биоэквивалентности или реже клинические исследования, в то время как за рубежом для ряда препаратов подобную оценку можно провести также на основании испытаний in vitro.
При всех методах оценки взаимозаменяемости генериков важной задачей является выбор препарата сравнения (референтного препарата). Наилучшим вариантом в качестве референтного препарата является выбор оригинального (инновационного) препарата, однако это не всегда представляется возможным. В различных нормативных документах (ВОЗ, FDA, Методические указания МЗСР РФ) приведены разные подходы к решению данной проблемы.
При решении вопроса об объеме экспертизы в рамках государственной регистрации необходимо найти компромисс между целесообразностью и достаточностью различных видов исследования. In vivo-in vitro корреляция (IVIVC) является эффективным прогностическим инструментом для оценки поведения лекарственных форм в условиях in vivo на основании профилей растворения препарата. Среди всех уровней корреляции наиболее достоверной для регуляторных целей является корреляция уровня А. Разработка такой корреляции тщательно нормируется и должна быть валидирована. Несмотря на то, что IVIVC в первую очередь применима к пролонгированным лекарственным средствам, ее также можно выявить и для лекарственных форм с немедленным высвобождением с учетом их положения в биофармацевтической классификационной системе. В настоящее время оценка in vivo-in vitro корреляции широко используется за рубежом.
Учитывая вышесказанное, а также вступление в силу нового закона «Об обращении лекарственных средств» данная работа, посвященная вопросам изучения воспроизведенных лекарственных средств с точки зрения их эффективности и безопасности, является крайне актуальной.
Цель исследования. Целью проведенного нами исследования являлось обоснование теоретических основ и разработка методических подходов по вопросу взаимозаменяемости воспроизведенных лекарственных средств и объему клинических исследований при их регистрации в Российской Федерации.
Для достижения указанной цели были поставлены следующие задачи:
1) дать характеристику Российского фармацевтического рынка на основе анализа номенклатуры воспроизведенных лекарственных средств;
2) провести анализ нормативно-правовой базы, регламентирующей объем экспертизы материалов воспроизведенных лекарственных средств при их государственной регистрации в РФ;
3) провести анализ документации, регламентирующей подходы к оценке эффективности и безопасности воспроизведенных лекарственных средств за рубежом, а также установлению взаимозаменяемости данных препаратов;
4) разработать методологические принципы изучения эквивалентности воспроизведенных препаратов с учетом гармонизации требований по вопросам эффективности и безопасности;
5) предложить системный подход к решению вопроса о взаимозаменяемости воспроизведенных лекарственных средств;
6) оценить возможности процедуры «биовейвер» при экспертизе материалов заявляемых к регистрации воспроизведенных лекарственных средств;
7) на основании изучения возможности применения in vitro-in vivo корреляции для препаратов различных фармакологических групп внести рекомендации для разработчиков и производителей лекарственных средств по ее использованию;
8) предложить список референтных препаратов для проведения исследований биоэквивалентности, сравнительной кинетики растворения и процедуры «биовейвер».
Научная новизна.
Впервые на основании анализа нормативных документов систематизированы подходы к оценке эквивалентности воспроизведенных лекарственных средств с позиции их взаимозаменяемости.
На основании проведенных исследований рекомендована процедура «биовейвер» при разработке нового воспроизведенного лекарственного средства.
По результатам проведенных исследований, сделано заключение о наличии корреляции уровня В для препаратов триметазидина (r = 0,97), индапамида (r – 0,94) и ципрофлоксацина (r = 0,96) и отсутствие данной корреляции для препаратов ибупрофена, что позволяет совершенствовать подходы к оценке их эффективности и безопасности.
Практическая значимость.
По результатам научных исследований и анализа нормативно-правовой базы, регламентирующей порядок экспертизы материалов эффективности и безопасности воспроизведенных лекарственных средств, заявляемых к государственной регистрации, разработаны предложения по доказательству их эквивалентности и оценке взаимозаменяемости, реализованные путем принятия нормативно-правовых актов федерального уровня.
Выполненная работа предлагает стандартизованный подход к оценке эффективности и безопасности воспроизведенных лекарственных средств на всех этапах их обращения – от создания до пострегистрационных исследований. Диссертационная работа позволяет: совершенствовать деятельность организаций и учреждений здравоохранения при оценке эффективности и безопасности лекарственных средств; использовать стандартизованные, гармонизированные процедуры при проведении клинических исследований воспроизведенных лекарственных средств.
Публикации. По теме диссертации опубликовано 38 печатных работ, в том числе 22 в изданиях, рекомендуемых ВАК РФ.
Апробация работы.
Апробация работы проведена на совместной научно-практической конференции Филиала «Клиническая фармакология» НЦ БМТ РАМН, кафедры клинической фармакологии и пропедевтики внутренних болезней Первого МГМУ им. (апрель, 2011 г.).
Основные результаты исследования были доложены на:
1) Российском национальном конгрессе «Человек и лекарство» (Москва, апрель г. г.);
2) Всероссийском совещании по вопросам государственного регулирования в сфере обращения ЛС и медицинской техники (Москва, октябрь 2004 г.);
4) Всероссийской фармацевтической конференции «Производители лекарственных средств на фармацевтическом рынке России: Стратегия развития» (Москва, март 2005 г.);
5) ХI Всероссийской конференции «Аптечная сеть России» (Москва, март 2006 г.);
6) Всероссийской конференции «Аптечный форум: от производителя до аптеки и потребителя» (Москва, апрель, 2006 г.);
7) VII научно-практической конференции «Биомедицина и Биомоделирование» (Москва, Московская обл. - Светлые горы, май 2011 г.);
8) 10th Congress of the European Association for Clinical Pharmacology and Therapeutics (EACPT) (Budapest, Hungary, June 2011).
Объем и структура диссертации.
Диссертационная работа состоит из введения, четырех глав, общих выводов и приложения. Работа изложена на 211 страницах машинописного текста, содержит 45 таблиц и 15 рисунков. Список литературы включает 192 источника (из них 123 иностранной) литературы.
Личный вклад автора.
Диссертантом лично проведен анализ рынка воспроизведенных лекарственных средств, систематизированы и критически оценены подходы к оценке эффективности и безопасности воспроизведенных препаратов в РФ и за рубежом, разработаны дизайн протоколов исследований, обработана первичная документация, в том числе индивидуальные регистрационные карты, проведены анализ и обобщение результатов, выполнена статистическая обработка, подготовлены нормативные документы, публикации и ряд сообщений на мероприятиях федерального и регионального уровней. Вклад автора является определяющим и заключается в непосредственном участии на всех этапах исследования: от постановки задач, до обсуждения результатов в научных публикациях и докладах и их внедрения в практику.
Работа с добровольцами и химический анализ образцов проводились сотрудниками Филиала «Клиническая фармакология» НЦ БМТ РАМН.
Соответствие диссертации паспорту научной специальности.
Научные положения диссертации соответствуют формуле специальности 14.03.06 – фармакология, клиническая фармакология, в том числе пунктам 7, 9, 19.
Основные положения, выносимые на защиту:
1) предложения по совершенствованию подходов к оценке эффективности и безопасности воспроизведенных лекарственных средств, с позиции их взаимозаменяемости;
2) концепция процедуры «биовейвер», для организаций осуществляющих разработку, производство и экспертизу лекарственных средств;
3) унифицированный подход к установлению in vitro-in vivo корреляции;
4) стандартизованные процедуры по оценке взаимозаменяемости воспроизведенных лекарственных средств.
ОСНОВНОЕ СОДЕРЖАНИЕ РАБОТЫ
Оригинальные и воспроизведенные лекарственные средства – реалии современного фармацевтического рынка
Большая часть лекарственных средств на современном фармацевтическом рынке является воспроизведенными (генерическими) препаратами [, 2008 г.] (Рис.1).
Среди препаратов, которые ежегодно регистрируются в России, отмечается значительно большее количество генериков, чем оригинальных препаратов. Отдельные оригинальные препараты имеют значительное количество воспроизведенных лекарственных средств. Так, оригинальное лекарственное средство «Вольтарен» (действующее вещество – диклофенак натрия) сегодня имеет 207 генериков, зарегистрированных к медицинскому применению. Также в РФ зарегистрировано около 150 генерических лекарственных средств эналаприла, около 100 - нифедипина, атенолола, ципрофлоксацина и порядка 50 – нитроглицерина, аспирина и парацетамола (причем количество комбинированных генерических последних двух лекарственных средств превышает 300). При этом следует отметить, что не всегда присутствующие на фармацевтическом рынке воспроизведенные лекарственные средства соответствуют требованиям спецификаций качества. В ряде стран с развитой системой контроля качества, эффективности и безопасности лекарственных средств, число генериков инновационного препарата не превышает 4-5.
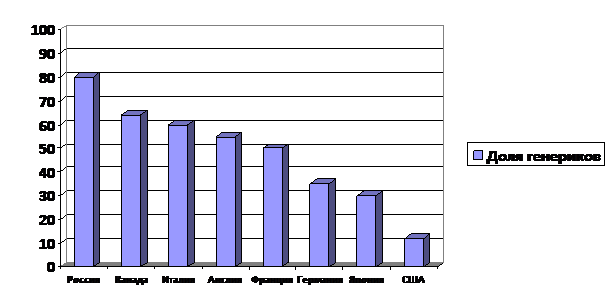
Рис.1. Доля генерического сектора фармацевтического рынка некоторых стран.
Таким образом, важной задачей Российского здравоохранения является обеспечение тщательного контроля качества воспроизведенных лекарственных средств и надлежащей документации, подаваемой в регуляторные органы при регистрации такого лекарственного средства.
Такая документация должна подтверждать, что генерическое лекарственное средство: соответствует стандартам GMP; соответствует требования спецификаций качества; соответствует по эффективности и безопасности инновационному (оригинальному) лекарственному средству.
Генерики, как и инновационные (оригинальные) препараты, должны отвечать общим требованиям, предъявляемым в рамках Общего (или единого) технического документа (CTD): эффективность, безопасность, качество. Качество оценивают фармакопейными методами, описанными в соответствующей нормативной документации (НД). При этом традиционно проводится анализ по трем основным направлениям: установление подлинности лекарственного препарата, оценка его чистоты и количественное определение. В НД могут быть включены также методики, оценивающие качество лекарственного препарата по некоторым специфическим, характерным для конкретной лекарственной формы параметрам. Например, средняя масса таблеток, однородность дозирования и другие.
В настоящее время на каждый генерик в России имеется свой НД как для зарубежных, так и для отечественных препаратов. Для последних, до вступления в силу нового Федерального закона «Об обращении лекарственных средств», — это были фармакопейные статьи предприятий (ФСП). При отсутствии единых (одинаковых или сопоставимых) требований каждый производитель, хотя и руководствуется общими фармакопейными статьями и ориентируется на зарубежные фармакопеи и существующую НД других производителей, может закладывать в свою НД нормы, которые в ряде случаев ухудшают качество препарата. Поэтому наличие большого количества генериков требует введения единых стандартов качества — фармакопейных статей (ФС), и уровень качества генерика должен быть не ниже того уровня, который определяется соответствующей ФС.
Основными проблемами, которые могут встречаться при использовании воспроизведенных лекарственных средств, можно считать следующие:
1. генерик произведен при действующем патенте на инновационный препарат (что является нарушением действующего законодательства);
2. производство генерика не соответствует требованиям GMP;
3. отсутствие эквивалентности инновационному препарату: фармацевтической; биологической; терапевтической;
4. стоимость незначительно ниже стоимости инновационного препарата;
5. профиль безопасности (частота развития нежелательных лекарственных реакций) у генерика выше;
6. при установлении эффективности и безопасности воспроизведенного и оригинального лекарственного средства:
- используются только исследования по оригинальному лекарственному средству без установления его эквивалентности генерику;
- нарушение протоколов исследований эквивалентности (число испытуемых и т. д.).
В связи с вышеизложенным, одной из основных задач клинической фармакологии является разработка подходов и протоколов исследования лекарственных средств, которые позволяли бы устанавливать соответствие (эквивалентность) эффективности и безопасности заявленного к регистрации воспроизведенного препарата инновационному лекарственному средству.
Несмотря на широкое использование данного понятия, «эквивалентности» генериков, как термина, не существует. Всемирная Организация Здравоохранения предлагает применять термин «взаимозаменяемость» (interchangeability) воспроизведенных лекарственных препаратов. Взаимозаменяемое генерическое лекарственное средство – это терапевтически эквивалентное генерическое лекарственное средство, которым можно заменить препарат сравнения в клинической практике. Видов «эквивалентности» воспроизведенных лекарственных средств выделяют несколько – терапевтическая, фармацевтическая, биологическая, а также так называемая «эквивалентность in vitro» (in vitro equivalence), введенная в употребление в документе «WHO Technical Report Series 937. WHO Expert Committee on Specifications for Pharmaceutical Preparations (2006). Annex 7. Multisource (generic) pharmaceutical products: guidelines on registration requirements to establish interchangeability».
Терапевтически эквивалентными лекарственные препараты могут считаться только в том случае, если они фармацевтически эквивалентны и можно ожидать, что они будут иметь одинаковый клинический эффект и одинаковый профиль безопасности при использовании пациентами в соответствии с указаниями инструкции по применению. Терапевтическая эквивалентность означает, что два препарата обеспечивают одинаковый терапевтический эффект и безопасность. Терапевтически эквивалентные лекарственные препараты должны отвечать следующим требованиям: иметь доказанную эффективность и безопасность; быть фармацевтически эквивалентными; быть биоэквивалентными; иметь сходные инструкции по применению; производиться в условиях стандарта GMP. Доказанную клиническую эффективность и безопасность устанавливают на основании клинических исследований. Клинические исследования лекарственных препаратов во всех развитых странах регламентируются законодательно. В России клинические исследования регламентируются следующими законами: Федеральным законом № 86 «О лекарственных средствах» (до 01.09.2010 г.), Федеральным законом № 61 «Об обращении лекарственных средств» (с 01.09.2010 г.), Национальным стандартом РФ ГОСТ-Р "Надлежащая клиническая практика», приказами Министерства здравоохранения и социального развития России и другими нормативными документами.
Оценка биоэквивалентности лекарственных средств является основным видом медико-биологического контроля воспроизведенных (генерических) лекарственных средств, не отличающихся лекарственной формой и содержанием действующих веществ от соответствующих оригинальных лекарственных средств. Исследования биоэквивалентности позволяют сделать обоснованные заключения о качестве сравниваемых препаратов по относительно меньшему объему первичной информации и в более сжатые сроки, чем при проведении клинических исследований. Тем не менее, такие исследование все равно требуют значительных временных и финансовых затрат, а также требуют вовлечения в исследование Этического комитета, поскольку такие исследования подразумевают участие здоровых добровольцев. Поэтому на протяжении последнего десятилетия неоднократно обсуждался вопрос о возможности заключения о взаимозаменяемости воспроизведенных лекарственных средств на основании исследований in vitr. В некоторых международных руководствах введено понятие регуляторной процедуры «биовейвер», в соответствии с которой определение взаимозаменяемости генерических лекарственных средств проводится на основании оценки их биофармацевтических свойств и эквивалентности in vitro (изучение сравнительной кинетики растворения), либо другими методами in vitro в качестве альтернативы исследованиям биоэквивалентности in vivo при их государственной регистрации.
Документ «WHO Technical Report Series 937. WHO Expert Committee on Specifications for Pharmaceutical Preparations (2006). Annex 7. Multisource (generic) pharmaceutical products: guidelines on registration requirements to establish interchangeability» дает четкое определение оценке эквивалентности in vitro – это испытание, предназначенное для оценки эквивалентности профилей растворения в трех средах со значениями рН 1,2, 4,5 и 6,8 исследуемого лекарственного средства и препарата сравнения или лекарственных средств одного производителя в различных дозировках. В Российской Федерации в настоящее время данное испытание не регламентируется законодательно, а испытания сравнительной кинетики растворения in vitro применяются только для оценки эквивалентности разных дозировок одного и того же генерического лекарственного средства.
Установление взаимозаменяемости воспроизведенных
лекарственных средств
Пути установления взаимозаменяемости воспроизведенных лекарственных средств
В совместном заявлении Международной Фармацевтической Федерации (FIP) и Международной Федерации Фармацевтических Производителей и Ассоциаций (IFPMA), принятом в 2000 г., указано, что замена оригинального лекарственного средства на воспроизведенное должна проводиться только в том случае, когда воспроизведенное лекарственное средство соответствует принятым международным стандартам, включая биоэквивалентность, с целью обеспечения качества всех препаратов на рынке.
Соответствие эффективности и безопасности, то есть терапевтического эффекта инновационного (или другого препарата сравнения) и генерического лекарственного средства обеспечивает их взаимозаменяемость в клинической практике, то есть терапевтическую эквивалентность. Эквивалентность может быть подтверждена: при сравнительных клинических исследованиях in vivo; при сравнительных исследованиях биоэквивалентности (фармакокинетических исследованиях) in vivo; при сравнительных фармакодинамических исследованиях in vivo; при сравнительных исследованиях in vitro (кинетика растворения) (в Российской Федерации не применяются для данной цели); в отдельных случаях оценка взаимозаменяемости не проводится.

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 |
