
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral

Рисунок 3. Роль калликреин-кининовой системы в поддержании уровня АД
Брадикинин - главный эффекторный пептид ККС, образуется из брадикининогена плазмы под влиянием калликреина и под влиянием кининпревращающего фермента в почках из кинина каллидина (лиз-брадикинин). Кинины быстро разрушаются ферментами (кининазой I и кининазой II, она же - АПФ) и живут от 17 до 24 сек. Действуя через брадикининовые рецепторы (в основном, В1 и В2), брадикинин вызывает: 1) сосудорасширяющее действие. Он действует непосредственно на стенку сосудов, а также через стимуляцию эндотелия к выделению NО, простациклина и простагландина Е; 2) снижает АД; 3) обладает натрийуретическим действием, так как стимулирует синтез в собирательных трубках и интерстициальных клетках почек простагландина Е, способствующего выраженному натрийурезу; 4) уменьшает антидиуретическое действие АДГ; 5) уменьшает высвобождение норадреналина из окончаний симпатических волокон и секрецию катехоламинов надпочечниками (рис. 3).
Показано снижение активности ККС у больных АГ. Возможно это связано с высокой активностью АПФ – кининазы II, который превращает брадикинин в неактивные пептиды. Не исключена и наследственная роль низкой активности ККС. В результате превалирует ответ антагонистических гипертензивных факторов (РААС, симпато-адреналовой системы и др.).
4. Эндотелиальная дисфункция.
Эндотелиоциты (ЭЦ) участвуют в регуляции тонуса сосудов, поскольку синтезируют наряду со многими важными веществами вазодилатирующие и вазоконстрикторные факторы. На состояние и функцию ЭЦ оказывают 3 основных группы факторов: 1.) изменение скорости кровотока (увеличение напряжения сдвига); 2) медиаторы тромбоцитов (серотонин, тромбин, АДФ); 3) биоактивные вещества, циркулирующие в крови и продуцируемые самими ЭЦ (катехоламины, АДГ, гистамин, брадикинин и др.) (рис. 4).
Основным вазодилататором, продуцируемым ЭЦ, является оксид азота - NО, кроме которого ЭЦ вырабатывают еще простациклин (простагландин I2) и др. вазодилататоры. NО образуется из L-аргинина при помощи эндотелиальной NО-синтетазы. Продукция NО бывает базальной и стимулированной. Базальная или постоянная продукция NО играет очень важную роль. Она обеспечивает оптимальную степень дилатации сосудов и препятствует вазоконстрикции. Уровень базальной секреции NО определяется пульсирующим характером кровотока по сосудам и силой воздействия потока движущейся крови на эндотелий, т. е. напряжением сдвига. Стимулированная продукция NО возникает в ответ на гипоксию, гуморальные вещества и механическую деформацию сосудов.
Основными стимуляторами секреции NО являются напряжение сдвига в сосудах, брадикинин и ацетилхолин (синтез NО увеличивают еще норадреналин, АДГ, гистамин, серотонин, тромбин, эндотелин). NО вызывает в гладкомышечной сосудистой клетке активацию гуанилатциклазы, рост цГМФ (циклического гуанозинмонофосфата), уменьшение Са ++ и вазодилатацию.
При ЭАГ продукция NО понижена. Возможно это связано с уменьшением активности NО-синтетазы (у больных выявлен полиморфизм гена NО-синтетазы), с повышением активности АПФ, разрушающего брадикинин – стимулятор синтеза NО, или с повышенной инактивацией NО свободными радикалами, возникающими при перекисном окислении липидов (ПОЛ).
Простациклин. Синтезируется ЭЦ из арахидоновой кислоты. Синтез стимулируется напряжением сдвига, брадикинином, веществом Р, тромбином, ИЛ-I, АТ-II, гипоксией. Является сильным антиагрегантом в отношении тромбоцитов. Расширяет сосуды за счет увеличения синтеза цАМФ (циклического аденозинмонофосфата) в гладкомышечных клетках и снижения их чувствительности к Са++. При ЭАГ продукция простациклина понижена.

РgI1, — простациклин,
АПФ — ангиотензинпревращающий фермент,
АТ-II — ангиотензин II,
ОПСС — общее периферическое сосудистое сопротивление
Рисунок 4. Роль дисфункции эндотелия в патогенезе артериальной гипертонии.
ЭЦ вырабатывают вазоконстрикторы – эндотелины (ЭТ-1,2,3,4), АТ-II (превращение АТ-I под влиянием АПФ), тромбоксан, простагландин Н2.
Наиболее сильный вазоконстриктор – ЭТ-1. Он образуется из своего предшественника – «большого эндотелина» - под влиянием эндотелинпревращающего фермента, обнаруженного внутри и на поверхности ЭЦ в разных органах и тканях. ЭТ разерушаются под действием различных протеаз, наибольшее значение имеют деамидаза и энкефалиназа. Синтез ЭТ-1 стимулируется АТ-II, АДГ, тромбином, модифицированными ЛПНП, гиперхолестеринемией, свободными радикалами, глюкозой, некоторыми факторами роста, гипоксией и напряжением сдвига. ЭТ-1 вызывает спазм сосудов. Он активирует в гладкомышечной клетке G-белки, фосфолипазу С, что приводит к повышению в клетке Са++ и сокращению клеток. ЭТ-1 стимулирует пролиферацию гладких мышц сосудов, оказывает положительное инотропное влияние на миокард и усиливает активность ренин-ангиотензиновой и симпато-адреналовой систем.
Обнаружены 3 типа ЭТ-рецепторов: А, В1 и В2, С. При взаимодействии ЭТ-1 с рецепторами А наблюдается вазоконстрикция и торможение синтеза NO.
При ЭАГ обнаружен повышенный уровень ЭТ-1 в крови. У крыс со спонтанной АГ также повышено содержание ЭТ-1. Некоторые исследователи полагают, что главным является повышенная продукция ЭТ-1 в сосудистой стенке независимо от содержания его в крови.
Таким образом, указанная эндотелиальная дисфункция – сниженная продукция вазодилататоров и повышенная – вазоконстрикторов, приводит к повышению ОПСС, АД, к ремоделированию артерий (гипертрофии, фиброзу) и снижению эластичности стенок артерий.
5. Нарушение депрессорной функции почек.
Почки вырабатывают гипотензивные простагландины, главным из которых является простагландин Е2 (PgE2). Он расширяет сосуды почек, улучшает почечный кровоток, повышает экскрецию натрия и воды, ингибирует синтез ренина и АТ-II. При АГ депрессорная система почек вначале активируется, затем истощается. Уменьшается продукция кининов, PgE2, что способствует проявлению действия вазопрессорных механизмов и стабилизации высокого АД.
Общая схема патогенеза ЭАГ представлена на рисунке 5.
ПАТОГЕНЕЗ ПОРАЖЕНИЯ ОРГАНОВ-МИШЕНЕЙ ПРИ ЭАГ
Высокое АД постепенно приводит к поражению органов-мишеней – сердца, сосудов, почек, головного мозга и сетчатки.
Гипертрофия сердца, прежде всего левого желудочка, является следствием перегрузки сердца давлением (постнагрузки), воздействия на миокард нейро-гуморальных факторов (катехоламинов, АТ - II, альдостерона и др.), наследственных нарушений (учитывая наследственную предрасположенность к ЭАГ).
Поражение периферических артерий заключается в их ремоделировании и протекает в две стадии: стадию функциональных изменений сосудов, обусловленную сосудосуживающими реакциями в ответ на трансмуральное давление и нейрогормональную стимуляцию, и морфологическую стадию, которая характеризуется структурным уменьшением просвета сосудов вследствие утолщения их медиального слоя. Сосуды становятся ригидными, так как в них увеличивается содержание матрикса, т. е. внеклеточного коллагена. Крупные артерии подвергаются атеросклеротическому поражению, а мелкие сосуды, артериолы и капилляры, редуцируются, в тканях развивается разряжение сосудистого русла с уменьшением объемного кровотока и развитием венозной гиперемии и стазов.
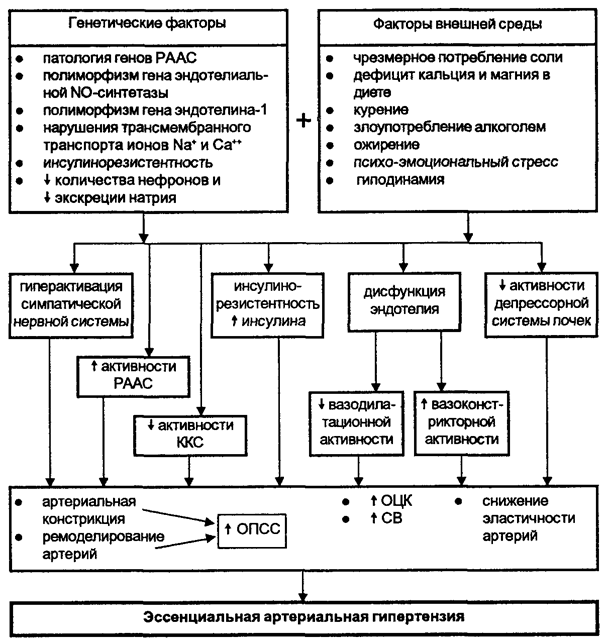
РААС - ренин-ангиотензин-альдостероновая система;
ККС - калликреин-кининовая система;
ОПСС - общее периферическое сосудистое сопротивление;
ОЦК - объем циркулирующей крови;
СВ - сердечный выброс.
Рисунок 5. Схема патогенеза эссенциальной артериальной гипертензич.
Поражение почек – гипертоническая нефропатия или гипертонический артериолосклеротический нефросклероз (первично сморщенная почка). Проходит ряд стадий. Вначале наблюдаются функциональные изменения сосудов почек. Повышение АД приводит к увеличению ультрафильтрации и выведения избытка натрия, способствуя нормализации АД. Для защиты от повышенного АД в почке возникает спазм приводящих афферентных артериол, постепенно приводящий к их ремоделированию. В последующих стадиях развивается гипертонус эфферентных сосудов. Артериосклеротические изменения сосудов, функциональная перегрузка клубочков и канальцев приводит постепенно к атрофии и диффузному склерозу почек.
В сосудах сетчатки при АГ развиваются те же механизмы поражения, как и в системе микроциркуляции.
Головной мозг поражается в результате ремоделирования как крупных, так и мелких сосудов.
ВТОРИЧНЫЕ АГ
Из вторичных или симптоматических АГ рассмотрим патогенез почечных АГ.
Заболевания паренхимы почек как правило приводят к развитию стойкого повышения АД, развитию ренопривных гипертензий. Патогенез их представляется следующим.
1). Активируется РААС и симпато-адреналовая система. 2). Задерживается натрий и вода в результате уменьшения массы действующих нефронов. 3). Снижается активность депрессорной системы почек, уменьшается синтез кининов, простагландинов, NO. 4). В почках активируются ПОЛ, продукты ПОЛ, свободные радикалы, угнетают синтез ЭЦ оксида азота, способствуют выделению вазопрессорных медиаторов из арахидоновой кислоты внутри клубочковых мембран. 5). Развивается дисфункция эндотелия и активация его сосудосуживающей функции. В норме NO вырабатывается ЭЦ почечных артериол, капилляров клубочка, областью плотного пятна. NO регулирует в почках кровоток, натрийурез, синтез и высвобождение ренина. При уменьшении выработки NO активируется синтез ренина, усиливается задержка натрия, возрастает протеинурия, прогрессирует гломерулосклероз, стабилизируется высокое АД. Кроме этого, увеличивается продукция эндотелина.
Заболевания почечной артерии (атеросклероз, тромбоз, эмболия и др.) приводят к развитию реноваскулярной АГ. Повышение АД развивается в результате 1) активации РААС из-за ишемии почки на стороне поражения и 2) развития оксидантного стресса. Механизм последнего сложен. Повышенный уровень АТ-II и АД активируют НАДН - и НАДФН-оксидазы в ЭЦ, гладких мышцах сосудов, макрофагах и др. клетках, что приводит к образованию перекиси водорода и супероксиданионрадикала. Они разрушают NO с образованием пероксинитрита (ООNО-). Пероксинитрит инициирует ПОЛ, повреждает ДНК, подавляет выработку энергии в митохондриях.
ПРИНЦИПЫ ТЕРАПИИ АГ
Для лечения АГ применяются следующие основные группы препаратов.
1. Диуретики. 2. Ингибиторы АПФ. 3. Антагонисты кальция. 4. Бета-адреноблокаторы. 5. Препараты последних поколений других групп (часто назначаются дополнительно к какому-то препарату из групп 1-2). Например, агонисты имидазолиновых рецепторов, периферические вазодилататоры, открывающие калиевые каналы, альфа-1-адреноблокаторы.
Рекомендуется ограничение в диете поваренной соли.
РЕКОМЕНДУЕМАЯ ЛИТЕРАТУРА
1. Физиология сердечно-сосудистой системы, Д. Морман, Л. Хеллер, 2000, «Питер».
2. Физиология человека, под редакцией Р. Шмидта и Г. Тевса, т.1 и 2, 1996, «Мир»
3. Гипертоническая болезнь, Е. Гогин, 1997, Москва.
4. Артериальная гипертензия, Б. Шулутко, Ю.Перов, 1993, С.-Пб.
5. Диагностика болезней внутренних органов: т.7. Диагностика болезней сердца и сосудов. Артериальная гипертензия, А. Окороков, 2003, М., Мед. лит.
6. Rhomantic interludes raise blood pressure, A. Somlyo, Nature, 1997, v.398, October: 908-911.
_________________________________________________________________
ОГЛАВЛЕНИЕ
стр. | |
Патофизиология артериальной гипертонии | 3 |
Прессорные механизмы | 7 |
Депрессорные механизмы | 9 |
Классификации АГ | 11 |
Этиологии АГ | 13 |
Патогенез АГ | 15 |
Патогенез поражения органов-мишеней при ЭАГ | 21 |
Вторичные АГ | 23 |
Рекомендуемая литература | 24 |
Патология сердечно-сосудистой системы. Часть III. Патофизиология артериальной гипертонии
Методические разработки для самостоятельной работы
студентов лечебного и педиатрического факультетов.
Под редакцией профессора , профессора
Составитель доцент
Гарнитура Таймс Объем 2,5 уч.-изд. л.
Тираж 600 экз.
ГБОУ ВПО РНИМУ, Москва, 117437, Островитянова, 1. 4223

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 |
