

Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
УДК 532.74
О ВЛИЯНИИ ГЕОМЕТРИИ ВОДОРОДНОГО МОСТИКА НА КОЛЕБАТЕЛЬНЫЕ СПЕКТРЫ ВОДЫ: ПРОСТЕЙШИЕ МОДЕЛИ ПОТЕНЦИАЛА H-СВЯЗИ
Институт химической кинетики и горения СО РАН
630090, Новосибирск, Российская Федерация.
Ключевые слова: жидкая вода, континуальная модель, водородная связь, флуктуационная теория, геометрия, потенциал, колебательные спектры.
АННОТАЦИЯ
Флуктуационная теория водородной связи (Н-связи) позволяет количественно реконструировать колебательные спектры жидкой воды и интерпретировать их в терминах континуальной модели. В развитие этой теории предлагается подход, позволяющий связать форму полос колебательных спектров со статистическим распределением геометрических параметров водородного мостика О-Н...О, порождённым флуктуациями локального окружения молекул в жидкости. Проверены две простейшие из предлагавшихся в литературе идей объяснения аномального уширения спектральных полос ассоциированных жидкостей: о доминирующем влиянии на частоту ОН-колебаний длины Н-связи, RO…O, либо угла её изгиба φ(Н-О...О). Показано, что каждая из них способна описать экспериментальные спектры и их температурную зависимость. Однако необходимая для этого взаимосвязь между геометрическими параметрами и частотой приводят к противоречию с рядом эмпирических фактов. Это говорит о недостаточности однопараметрических потенциалов для количественного описания водородной связи в жидкостях. Вместе с тем, предлагаемый подход может быть применён для конструирования и проверки более сложных потенциалов, включающих все геометрические параметры водородного мостика, влияющие на энергию Н-связи.
ВВЕДЕНИЕ
Проблема структуры жидкой воды и интерпретации её колебательных спектров имеет более чем вековую историю, ей посвящены тысячи научных публикаций (см. прекрасный обзор [a]). На сегодня доминирующими являются две группы моделей строения воды. Смешанная (mixture) модель исходит ещё к Рентгену [b] и в разных модификациях представляет воду как смесь различных ассоциатов (мономеров, димеров, малых кластеров и т. д.). Обязательным атрибутом смешанной модели являются разорванные водородные связи (см., например, [c, d]). Непрерывная (continuum) модель, напротив, трактует её скорее как «полимер» с флуктуирующей во времени структурой. Его строение определяет непрерывная трёхмерная сетка водородных связей, различающихся своими геометрическими параметрами, энергией и частотами колебаний вовлечённых осцилляторов. К основоположникам этой модели можно отнести Бернала и Фаулера [e] и Лэнгмюра, образно сравнившего океан с одной макромолекулой. Историю и современную трактовку этой модели можно найти, например, в [f, g].
Математический аппарат смешанной модели гораздо более прост; возможно, поэтому её больше любят химики и экспериментаторы. Так, в рейтинг первой десятки мировых научных достижений 2004 года [1] включена работа интернационального коллектива
-=-=-=-=
*****@***nsc. ru
учёных [2], исповедующих смешанную модель. Однако основной вывод авторов о том, что половина водородных связей в воде разорвана и продолжает рваться с повышением
температуры, противоречит как ряду надёжно установленных экспериментальных фактов, так и результатам компьютерного моделирования. В частности, он не согласуется с формой колебательных спектров и её температурной зависимостью. Причина заключается, по-видимому, в том, что совершенно правильные результаты эксперимента (уменьшение средней энергии водородных связей в расчёте на молекулу с ростом температуры) авторы интерпретируют как вызванный нагреванием разрыв части водородных связей, тогда как оставшиеся связи по-прежнему столь же сильны, как во льду. Другой альтернативы в канонической смешанной модели нет: нагрев должен приводить к перераспределению концентраций различных сортов объектов с фиксированными свойствами.
В терминах непрерывной модели эти же эксперименты качественно объясняются не разрывом отдельных, а ослаблением всех Н-связей воды, составляющих единый статистический ансамбль. Более того, теория, основанная на непрерывной модели, уже позволила количественно описать форму экспериментальных спектров комбинационного рассеяния [3,4] и инфракрасного поглощения [5,6] от 0 до 200оС, объяснить причину их существенно разного поведения, и рассчитать вклад водородных связей в основные термодинамические функции [7]. Следует отметить, что эта теория является чисто статической, т. е. не рассматривает временную динамику изучаемого ансамбля молекул. Однако новаторские работы в этой области, успешно объединившие возможности и средства молекулярной динамики, квантовой химии и фемтасекундной лазерной спектроскопии [i, j] подтвердили адекватность статического приближения. В частности, оказалось, что основой аномально широких полос в спектрах воды являются именно статические контуры [i], а динамические эффекты (типа спектральной диффузии, давно известной в ЯМР [Kubo], но не обсуждаемой ранее в оптике) лишь несколько модифицируют их форму [i, j]. К сожалению, выполненные в этих работах расчёты пока доступны лишь для очень малых ансамблей молекул (порядка сотни), требуя при этом огромных компьютерных ресурсов (тысячи часов процессорного времени). Вместе с тем, и теоретический анализ вновь тщательно измеренных спектров комбинационного рассеяния воды однозначно свидетельствует в пользу непрерывного статистического распределения энергий квазитетраэдрически координированных водородных связей её молекул [h]. Всё это делает целесообразным дальнейшее развитие континуальной модели в её статическом описании путём установления взаимосвязи между геометрическими, энергетическими и спектроскопическими характеристиками водородных связей в воде. В настоящей работе мы строго решаем задачу, какой должна быть эта взаимосвязь, чтобы количественно описать температурную эволюцию экспериментальных спектров на основе двух простейших из обсуждавшихся в литературе объяснений природы широкого распределения колебательных частот воды в терминах статистики геометрических параметров водородных связей в жидкости.
СТАТИСТИКА ЭНЕРГИЙ Н-СВЯЗЕЙ И ЕЁ ПРОЯВЛЕНИЯ В ФОРМЕ СПЕКТРАЛЬНОГО КОНТУРА
Согласно предложенному [8] алгоритму реализации флуктуационной теории водородной связи, статистическое распределение частот колебаний ОН-групп молекул воды при любой температуре Т считается больцмановским
P(ν,T) = Q-1(T)W(ν)exp[-E(ν)/(kbT)] (1)
и определяется как энергией конкретной Н-связи E(ν), так и её вырожденностью W(ν). Здесь Q(T) – статистический интеграл в общепринятом понимании (в (1) он нормирует распределение P(ν,T) по площади к единице), kb – постоянная Больцмана. Как видим, частота νOH является единственным параметром, через который выражаются все остальные. Интересно, что и авторы новейших работ [h-j] на основе разных методов (квантовохимическая оптимизация, анализ структуры множества молекулярно-динамических моделей) пришли к сходному выводу, что частота νOH определяется единственной величиной – проекцией напряжённости электрического поля на направление OH-группы в месте нахождения протона. Таким образом, в двух совершенно различных подходах к интерпретации спектров выявлен единственный параметр (энергия конкретной водородной связи либо напряжённость электрического поля, наведенного на ней соседями), который определяет частоту OH-колебаний.
В [k,4] было показано, что все три функции, входящие в (1), могут быть вычислены из колебательных спектров полудейтерированной воды HOD при нескольких температурах (рис. 1). Реконструированные на их основе с помощью формулы (1) спектры описывают
 |
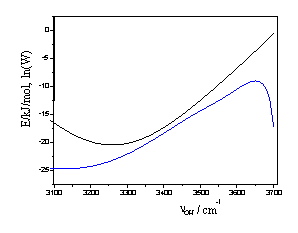

Рис. 1 Рис. 2
Рис. 1. Частотная зависимость функций E(ν) и логарифма W(ν) формулы (1), вычисленная в [k] из ИК-спектров воды на интервале 10 – 200оС. Вид статистического интеграла Q(T) см. также в [k].
Рис. 2. Экспериментальные (точки) и реконструированные в [4] по формуле (1) (сплошные кривые) спектры ОН-колебаний жидкой воды при постоянной плотности в изотропной составляющей комбинационного рассеяния. T= 10, 50, 90, 200oC (слева направо).
эксперимент во всём исследованном интервале температур лучше, чем любая другая из существующих ныне теорий (рис.2). К сожалению, формула (1) является по сути феноменологической. Она не содержит структурных параметров: и энергия, и вырожденность конфигураций водородного мостика напрямую связаны с частотой ν. В частности, всевозможные комбинации различных длин связи и углов её изгиба, дающие в результате одинаковую энергию Е, дают вклад в одну и ту же частоту ν(Е). А доля таких в принципе возможных конфигураций от их общего количества определяет вырожденность W(ν), которая, казалось бы, должна содержать структурную информацию о многообразии конфигураций Н-связей в воде. Удивительно, что столь простая гипотеза оправдалась с высокой степенью точности.
Однако хотелось бы попытаться выйти за рамки феноменологии и выяснить конкретную взаимосвязь геометрических параметров конфигурации водородного мостика с соответствующей им частотой в колебательном спектре, νOH, а тем самым и с энергией водородной связи. Как ни странно, в принципе это можно сделать, не выходя из рамок спектроскопии: путём анализа функции W(ν) формулы (1). Пусть G - совокупность всех геометрических параметров фрагмента O-H…O, влияющих на частоту ν, а W(G) – мера множества конфигураций, порождающих данную частоту. Тогда для сохранения дифференциала площадей в конфигурационном и частотном представлении W(G)dG должна равняться W(ν)dν, или в интегральном представлении
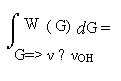

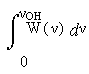
 (2)
(2)
То есть интеграл уже известной нам функции W(ν) (рис. 1) от нуля до любого наперёд заданного значения νOH должен равняться интегралу по контуру искомой функции W(G), в котором область интегрирования охватывет все совокупности значений геометрических параметров G, соответствующих частотам ν ≤ νOH. В случае, если множество G содержит несколько геометрических параметров, интегральное уравнение (2) приводит, вообще говоря, к некорректной обратной задаче, требующей специальных подходов. В настоящей работе мы рассмотрим простейшие случаи, когда энергия Н-связи зависит только от одного геометрического параметра. К таковым относятся две широко известные идеи объяснения аномального уширения спектральных полос воды: о зависимости частоты νOH от длины Н-связи, RO…O [9], либо от угла её изгиба φ(Н-О...О) [10], соответственно. (Кстати, и в новых работах [h, j,k] основной причиной различия частот разных ОН-осцилляторов называется вариация длин водородных связей, с которой мы и начнём).
Геометрию водородного мостика O-H…O мы будем характеризовать длиной связи R= RO…O и углом изгиба (отклонением H-связи от линейной) φ = φ(Н-О...О). Тогда чисто геометрическая (без учёта энергетики) вероятность для протонодонорной молекулы Н2О встретить в жидкости атом кислорода протоноакцепторной молекулы на расстоянии R±dR/2, расположенный под углом φ±dφ относительно направления O-H группы определится дифференциалом конического сечения шарового слоя:
W(R,φ) dRdφ = 4πR2 sin(φ)dRdφ. (3)
Роль энергии водородной связи с такой конфигурацией будет отражена в полной вероятности реализации P(R,φ,T), аналогичной формуле (1), экспонентой exp[-E(R, φ)/kT]. Таким образом, при однопараметрическом представлении потенциала водородной связи нам предстоит найти зависимости E(R) и E(φ) соответственно, удовлетворяющие уравнению (1), т. е. описывающие с его помощью температурную эволюцию экспериментальных спектров (рис. 2).
РАДИАЛЬНАЯ ЗАВИСИМОСТЬ
Пусть частота νOH однозначно определяется только длиной водородного мостика RO…O. Подобная зависимость
νOH(см-1) = 3707 – 2.222 *107*exp(-3.925 R), R(Å). (4)
давно известна для кристаллов [11,12], где ввиду идентичности всех Н-связей данного типа спектральные линии достаточно узки, и их частоты без труда сопоставляются соответствующим кристаллографическим расстояниям RO…O. Формулы (2),(3) позволяют найти аналогичную зависимость для жидкости, необходимую для описания температурной трансформации экспериментальных спектров. Поскольку в этом варианте мы не учитываем влияние угла φ, то из формулы (3) следует W(R) ~ R2. Тогда формула (2) приобретает конкретный вид:
J(νOH ) ≡ 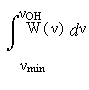
 =
= 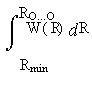
 = (R3O...О-R3min) / (Rlim3 - R3min) . (5)
= (R3O...О-R3min) / (Rlim3 - R3min) . (5)
Здесь Rmin - дистанция максимально возможного сближения молекул воды в жидкости, а νmin - соответствующая этому плотнейшему контакту частота ОН-колебаний, 3100 см-1, с которой начинается статистическое распределение P(νOH ) [4]. Тогда из формулы (4) следует Rmin = 2.677 Å. Третий параметр в формуле (5), Rlim, есть предельное межмолекулярное расстояние RO…O, при котором водородная связь О-Н...О между данной парой молекул ещё сохраняется (общепринятые оценки от 3.1 до 3.3Å ). Физический смысл предельной длины Н-связи для жидкой воды состоит в том, что в плотной жидкости, в отличие от димера в газе, чрезмерно удалившегося партнёра тут же заменит более близкий сосед со стороны (переключение связи на него). Знаменатель правой части формулы (5) играет роль нормировочного множителя, превращающего её в единицу при предельном значении νOH = νu = 3707 см-1 (частота колебаний свободной ОН-группы) в левой части уравнения. Поведение интеграла J(νOH ) функции W(ν), на котором основаны все дальнейшие расчёты, изображено на рис.3.
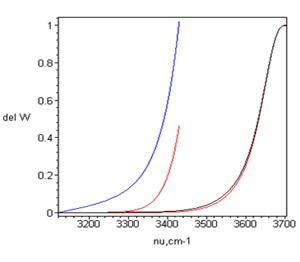
Рис. 3. Основа дальнейших расчётов: левая часть уравнения (5), как первообразная функции W(ν) (функция верхнего предела интегрирования). Левая кривая умножена на 100.
Решение уравнения (5)
RO…O(νOH ) = [R3lim+( R3lim - R3min) J(νOH )])] 1/3 (6)
определяет связь между величинами RO…O и νOH (кривая 1 на рис. 4а), удовлетворяющую формуле (1) после пересчёта функции E(ν) посредством (6) в E(RO…O). Будучи точным решением, такая зависимость E(R) (рис. 4б) совместно с W(R) = 4πR2 автоматически описывает форму экспериментальных спектров и их температурную зависимость, изображённую на рисунке 2. Однако эта зависимость очень сильно отличается от известной для кристаллов корреляционной кривой νOH (RO…O) [12] (кривая 2 на рис. 4а).
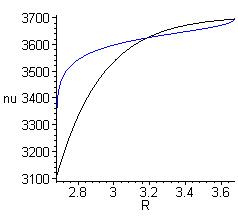
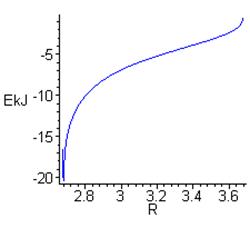
Рис. 4. а) Зависимость частоты ОН-колебаний от длины водородного мостика: синяя кривая - согласно формуле (6), чёрная – эмпирическая корреляция [12].
б) Зависимость энергии Н-связи от длины водородного мостика RO…O, вычисленная из E(ν) на рис. 1 с помощью формулы (6).
Самой яркой особенностью поведения энергии (рис. 4б) является то, что плавный минимум на кривой E(ν) (см. рис.1) в координатах E(R) превращается в очень узкий пик вблизи RO…O = 2.7 Å (длина Н-связей в гексагональном льду). Причина заключается в том, что полученная зависимость ν(R) конформно преобразует очень широкий интервал спектра между 3100 и 3500 см-1 в малую окрестность дистанций вблизи 2.7 Å (рис. 4а). В результате вычисленное на её основе распределение межатомных расстояний
P(RO…O ,T) = Q-1(T)W(RO…O)exp[-E(RO…O)/(kbT)] (7)
(рис. 5в) разительно отличается от 1-го максимума экспериментальных ФРР жидкой воды [13] и результатов компьютерного моделирования [14], гораздо более широкого и центрированного правее RO…O = 2.85 Å. Отметим, что существование минимума в зависимости E(ν) является необходимым условием корректного описания низкочастотного крыла спектров при низких температурах [4].
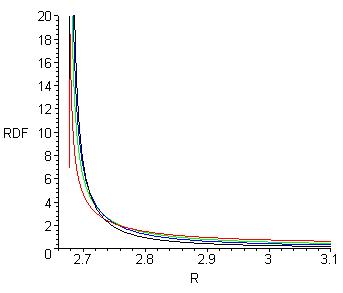
Рис.5в. Распределение длин водородной связи P(RO…O, T), вычисленное по формуле (7).
Чёрная кривая – 0оС, синяя – 50, зелёная – 100 и красная - 200 оС.
УГЛОВАЯ ЗАВИСИМОСТЬ
Пусть теперь частота νOH определяется не длиной водородного мостика RO…O, а, согласно гипотезе [10], углом её изгиба φ(Н-О...О). Тогда из (3) следует W(φ) ~ sinφ, а формула (2) редуцируется к уравнению
|
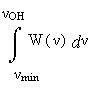
 =
= 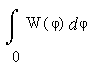
 = {1 – cos[φ(νOH)] } / [1 – cos(φlim)] . (8)
= {1 – cos[φ(νOH)] } / [1 – cos(φlim)] . (8)
Здесь мерой множества углов с данным значением φ является длина окружности образующей конуса с углом φ, а предельный угол φlim – угол разрыва (переключения) водородной связи имеет тот же смысл, что и Rlim в формуле (5) (по разным оценкам от 60 до 90о). В результате искомая связь частоты νOH и угла φ определяется соотношением
φ(νOH) = arcos{1- [1 – cos(φlim)] J(νOH )} (9)
и изображена кривой 1 на рис. 6. (Обратную зависимость νOH(φ) описать аналитически весьма сложно). Вычисленные в результате распределения углов
P(φ,T) = Q-1(T)W(φ)exp[-E(φ)/(kbT)] (10)
(рис. 7а) не столь сильно, как в случае радиальной зависимости, но весьма существенно отличаются от результатов компьютерного моделирования [15].
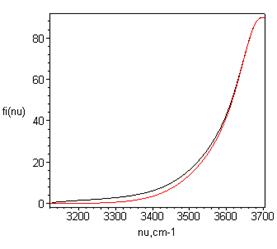
Рис.6.
Связь угла изгиба водородной связи φ(Н-О...О) и частоты νOH, следующая из выражения (9) и позволяющая столь же хорошо, как формула (1), описать трансформацию колебательных спектров
Они качественно верно отражают их форму и температурную зависимость. Однако максимумы рассчитанных при различных температурах распределений в 2-3 раза смещены к малым углам по сравнению с «компьютерными», а полуширины во столько же раз меньше. Достоверными экспериментальными данными об этих распределениях мы, к сожалению, не располагаем.
Соответствующая формуле (9) зависимость энергии Н-связи от угла её изгиба изображена на рис. 7б. Минимум кривой вблизи 5о формально является прямым следствием аналогичного минимума кривой E(ν). В терминах единственного значимого параметра (угла) оптимальность несколько изогнутой водородной связи можно объяснить стерическим фактором взаимодействия двух Н-связей, формируемых ОН-группами одной молекулы, как это имеет место, например, во льдах. Полезно отметить, что численное значение предельного угла φlim непринципиально: оно определяет лишь масштаб соответствующей оси. Так, замена φlim = 90о например, на 45о приведёт только к «растягиванию» кривых на рис. 6 и 7 в два раза: достаточно на соответствующей оси вместо 90о поставить 45о. Это, кстати, значительно улучшит согласие с результатами компьютерного моделирования.
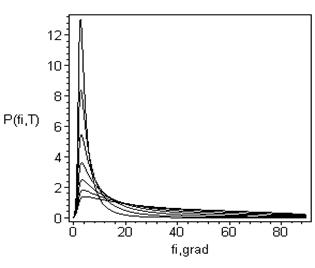
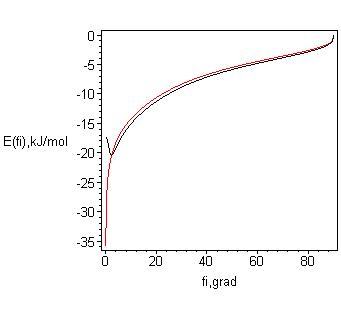
Рис. 7.
а) Следующие из формул (9-10) распределения угла изгиба (отклонения от линейности) Н-связи в жидкой воде. ). Т = -100, -50, 0, 50, 100, 150, 200 C (сверху вниз).
б) Зависимость энергии водородной связи от её изгиба Е[φ (ν)]
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Проведенные расчёты показывают, что обе предлагавшиеся в литературе альтернативные гипотезы о причинах аномально большого уширения спектров жидкой воды формально позволяют описать распределения частот в экспериментальных спектрах во всём ныне доступном интервале температур. Однако требующиеся для этого зависимости ν(RO…O) и ν(φ) противоречат известным эмпирическим корреляциям между соответствующими величинами, а вычисленные на их основе распределения межмолекулярных расстояний P(RO…O) и P(φ) весьма далеки от результатов анализа компьютерных моделей и экспериментальных ФРР воды. Особенно плохо в этом смысле пренебрежение ролью изгиба водородной связи, т. е. попытки объяснить спектры воды статистикой межмолекулярных расстояний RO…O (типа [9, ].
Вместе с тем, продемонстрированный здесь подход может быть применён для конструирования более сложных потенциалов, включающих одновременно все геометрические параметры водородного мостика, влияющие на энергию Н-связи. При этом использование эмпирических корреляций (типа формулы (4)) поможет найти корректное решение многопараметрического интегрального уравнения (2) путём реконструкции такого поведения интеграла J(νOH ), которое обеспечивает согласие искомой связи частоты и геометрических параметров с поведением экспериментальных спектров.
Важно отметить, что речь здесь идёт о потенциале именно водородной связи, т. е. о зависимости её энергии от расстояния между образующими эту связь молекулами и их взаимной ориентации. Такой потенциал может оказаться очень полезным, например, при анализе больших компьютерных моделей воды с целью выявления в них водородных связей, построения их сеток, количественного описания локальных свойств сеток и изучения топологии в целом. Вместе с тем, для непосредственного использования в молекулярно-динамических или Монте-Карло расчётах такой потенциал должен быть дополнен не учтёнными в нём взаимодействиями и, в первую очередь, взаимным отталкиванием партнёров по связи и их взаимодействием с прочими молекулами. Однако искомое нами «ядро» полного потенциала позволит очень легко вычислять для каждой компьютерной модели соответствующие ей колебательные спектры, независимо тестируя тем самым адекватность полученных результатов опыту.
БЛАГОДАРНОСТИ
Автор искренне благодарен , и за многочисленные обсуждения и полезные замечания.
Работа была поддержана грантами РФФИ 04-03-32560 и 07-03-00503-а.
ЛИТЕРАТУРА
[a]. // Журн. структур. химии. – 2006. – 47, (Приложение). – c. 5 – 35.
[b]. Röntgen W. C. // Ann. Phys. Chem. N. F. – 1891. – XLV. – S. 91 – 97.
[c]. Walrafen, G. E. // J. Chem. Phys. – 1968.- 48, - P. 244-.
[d]. Luck
[e] Bernal J. D., Fowler R. H. // J. Chem. Phys. – 1933. – 1. – P. 515 – 548. Русский перевод: Бернал Дж., Фаулер Р. // Успехи физ. наук. – 1934. – 14. – С. 586 – 644.
[f] И. Структурные модели жидкостей. – Новосибирск: Изд-во Новосиб. ун-та, 1983. – 83 с.
[g]= [3] Efimov Yu. Ya., Naberukhin Yu. I. // Faraday Discuss. Chem. Soc. – 1988. -85. - P. 117- 123.
[h] Smith J. D., Cappa Ch. D., Wilson K. R., et. al. // Proc. Natl. Acad. Sci. USA.- 2005.-102; -P. 14171-14174.
[i] Corcelli S. A., Skinner J. L. // J. Phys. Chem. - A 2005, -109, - P. 6154-6165
[j] Torii H. // J. Phys. Chem. - A 2006, -110. – P. 9469-9477.
[Kubo] Kubo R. // Adv. Chem. Phys. - 1969, 15, – P. 101- .
[k] Lawrence C. P., Skinner J. L. // J. Chem. Phys. – 2003. – 118, N 1. – P. 264 – 272.
[1]. Breakthrough of the Year («Прорывные» работы Года). // Science. – 2004. -306. - P. 2017.
[2] Wernet Ph., Nordlund D., Bergmann, U., et. al. // Science. - 2004. -304. - P. 995-999.
[3] Efimov Yu. Ya., Naberukhin Yu. I. // Faraday Discuss. Chem. Soc. – 1988. -85. - P. 117- 123.
[4] Efimov Yu. Ya., Naberukhin Yu. I. // Molecular Physics. – 2003. -101. - P. 459-468.
[5] Georgiev,
[6] Efimov Yu. Ya., Naberukhin Yu. I. // Molecular Physics, v.102, pp. 1407-1414 (2004).
[7] Efimov Yu. Ya., Naberukhin Yu. I. // Spectrochimica Acta A – 2005. - 61/8 - P. 1789-1794.
[8] Жуковский А.П. // Журн. Структурной химии. – 1976. – 17. – С. 931 – 932.
[9] Wall and Hornig
[10] (last)
[11] Корр. Кривая старая
[12] Корр. Кривая наша
[13] ФРР воды (Нартен)
[14] ФРР (МД, МК)
[15] P(φ) (МД, МК)
-=-=-=-=
ГГМ:
Сверхбыстрая лазерная ИК спектроскопия используется и для выяснения более тонких аспектов динамики водородных связей [138]. Работая на разных частотах в области валентных колебаний (~3500 см–1), авторам удалось проследить за зависимостью расстояний rOO от времени для связей слабых, сильных и средней силы. Если возбуждающее излучение соответствует 3510 см–1, то изучается сокращение связей с начальной длиной 3 Å до средней длины около 2,86 Å в течение примерно 2 пс (связь сокращается вдвое приблизительно за 0,5 пс). Если возбуждающее излучение было 3340 см–1, то следили за процессом удлинения связей от 2,8 до 2,86 Å. При возбуждающем излучении 3420 см–1 длина связей не зависела от времени и была равна среднему значению 2,86 Å.
Применение аналогичных методов быстрой ИК спектроскопии позволило установить корреляцию между мгновенной частотой колебания валентной связи O—H и длиной водородной связи (rOO), а также зависимость этой мгновенной частоты от количества водородных связей,
в которых данная молекула участвует [139].
138. Bratos S., Leicknam J.-Cl., Pommer S., Gallot G. // J. Mol. Struct. – 2004. – 708. – P. 197 – 203.
139. Lawrence C. P., Skinner J. L. // J. Chem. Phys. – 2003. – 118, N 1. – P. 264 – 272.
ПОДПИСИ К РИСУНКАМ
Рис.1. Частотная зависимость функций E(ν) и логарифма W(ν) формулы (1), вычисленная в [k] из ИК-спектров воды на интервале 10 – 200оС. Вид статистического интеграла Q(T) см. также в [k].
Рис.2.
а) Экспериментальные (точки) и реконструированные в [4] по формуле (1) (сплошные кривые) спектры ОН-колебаний жидкой воды в изотропной составляющей комбинационного рассеяния. T= 10, 50, 90, 200oC (слева направо).
б) То же для ИК-спектров (см. [6]).
Рис.3. Основа дальнейших расчётов: левая часть уравнения (4), как первообразная функции W(ν) (функция верхнего предела интегрирования). Левая кривая умножена на 100.
Рис.4.
а) Зависимость частоты ОН-колебаний от длины водородного мостика: синяя кривая - согласно формуле (5), чёрная – эмпирическая корреляция [11].
б) Зависимость энергии Н-связи от длины водородного мостика RO…O, вычисленная из E(ν) на рис. 1 с помощью формулы (5).
Рис.5.
Распределение межмолекулярных расстояний P(RO…O, T) в жидкой воде при нескольких температурах, вычисленное по формуле (5).
Чёрная кривая: 0оС, синяя: 50, зелёная: 100 и красная: 200 оС.
Рис.6.
а) Связь угла изгиба водородной связи φ(Н-О...О) и частоты νOH, следующая из выражения (8) и столь же хорошо, как формула (1), описывающая трансформацию колебательных спектров
б) Следующие из формул (8-9) распределения угла изгиба (отклонения от линейности) Н-связи в жидкой воде. ). Т = -100, -50, 0, 50, 100, 150, 200 C (сверху вниз).
Рис. 6Х. Зависимость энергии водородной связи от её изгиба Е[φ (ν)]
Рис. 7. Сплошная кривая: W(R), полученная из W(ν) (см. рис. 1) по формулам (10), (11).
Штрих: равный ей по площади отрезок кривой W(R)=R2 (см. выше).
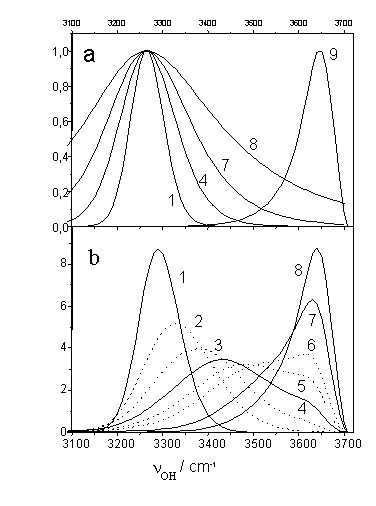 |
Fig6Ram. doc -> графический объект MS Office
Figure 6. The origin of temperature evolution of a statistical contour and its extrapolation to a wider temperature range. (a) The mechanism of statistical contour formation. Two cofactors determining the P(n) shape according to (1): the Boltzmann exponent at T= -173, 0, 200, and 1000oC – curves 1,4,7,8 at the left – and the function of state degeneracy W(n) – curve 9 at the right (all of them are normalized in height to unity). (b) Statistical contours being the product of above cofactors calculated for -173, -100, -50, 0, 50, 100, 200, and 1000oC – curves 1-8, respectively. The contours are normalized in area to unity.
