

Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
1) если ∆R/R1 < 4‑5 , то изоморфные замещения непрерывны при всех температурах;
2) при 4‑5 < ∆R/R1 < 14‑16 % непрерывный изоморфизм возможен только при высоких температурах; с понижением температуры происходит распад изоморфных смесей;
3) при 14‑16 < ∆R/R1 < 20‑25 % изоморфизм становится ограниченным даже при самых высоких температурах;
4) при ∆R/R1 > 20‑25 % заметных замещений не наблюдается. Хотя близость размеров замещающих друг друга атомов или ионов ‑ очень важное и обязательное условие, оно не является достаточным. Другим важным эмпирическим обобщением являются представления о направленности («полярности») изоморфизма. Так, в случае изовалентных замещений правило полярности Гольдшмидта гласит: ион с меньшим радиусом будет входить в общую кристаллическую структуру легче, чем ион с большим радиусом, занимающий ту же позицию. В случае гетеровалентных замещений действует другое правило полярности Гольдшмидта: ион с большим зарядом входит в кристалл легче, чем ион с меньшим зарядом, занимающий ту же кристаллографическую позицию.
Главные правила изоморфизма являются следствием факторов, которые вызывают повышение энергии кристалла в результате геометрической (структурной) или электронной (химической) деформации твердого раствора при образовании его из чистых кристаллов. Структурная деформация происходит из-за различия параметров (∆R), а электронная ‑ из-за различия в строении электронных оболочек замещающих друг друга атомов и возникающего вследствие этого «возмущения».
Твердые растворы при изоморфном замещении часто хорошо описываются как строго регулярные, для которых молярная энтропия растворения аналогична энтропии образования идеального раствора:
![]() (2)
(2)
(хi, — мольная доля i-го компонента; к — константа Больцмана; NA — число Авогадро), а молярная энтальпия смешения является квадратичной функцией состава:
![]() (3)
(3)
где Q — коэффициент.
Критерием адекватности той или иной модели твердого раствора (позволяющей оценить коэффициент Q) является ее способность предсказать положение (в координатах температура ‑ концентрация) линии купола распада этого раствора. Эта линия в случае регулярного раствора описывается уравнением (d(∆gсм)/dx1 = 0; gсм ‑ молярная энергия Гиббса процесса смешения:
![]() (4)
(4)
причем купол распада может наблюдаться только в случае Q > 0. Активность i-го компонента бинарного твердого раствора может быть выражена уравнением
![]() (5)
(5)
Квадратичная концентрационная зависимость энтальпии смешения может быть легко получена в приближении локализованных парных межатомных взаимодействиях (например, для случая ковалентного кристалла) и совершенно неупорядоченного распределения компонентов твердого раствора. Пусть твердый раствор компонентов А и В с концентрациями х1 и х2 соответственно характеризуется энергиями парных взаимодействий ЕАА, ЕВВ и ЕАВ, тогда энтальпия смешения может быть записана в виде
![]() (6)
(6)
где пАA, пвв, пАВ — концентрации соответствующих пар атомов; Z ‑ координационное число.
Поскольку каждый атом твердого раствора имеет Zx1 соседей А и Zx2 соседей В, концентрации пар атомов равны
![]()
![]()
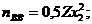
![]()
Подставляя эти значения в уравнение (6), получаем
![]() (7)
(7)
Из уравнения (7) следует, что меньшее значение энергии взаимодействия разноименных атомов в сравнении с одноименными препятствует образованию твердого раствора (или химического соединения), а большее ‑ способствует. В первом случае говорят о склонности системы к микрорасслаиванию (∆Нсм > 0), во втором случае ‑ о склонности системы к микроупорядочению (∆Нсм < 0). Информация о склонности твердого раствора микрорасслаиванию или микроупорядочению может быть получена методом рентгеновской дифракции из анализа интенсивности фона между брегговскими рефлексами. Некоторые данные о отнесении исследуемой системы к тому или иному случаю можно получить из анализа концентрационной зависимости параметров элементарной ячейки. Положительные отклонения от правила Вегарда (линейной концентрационной зависимости параметров элементарной ячейки) обычно свидетельствуют о склонности твердого раствора к микрорасслаиванию при понижении температуры, а отрицательные ‑ к микроупорядочению.
Выражение, соответствующее случаю регулярного раствора, может быть получено и для кристаллов с ионной связью, причем параметр Q может быть выражен через индивидуальные характеристики ионов. Рассмотрение энтальпии смешения гомовалентных твердых растворов замещения как изменения энергии связи ионного кристалла в зависимости от межатомного расстояния (с условием выполнения правила Вегарда) с последующим разложением в ряд Тейлора по параметру ∆R/R в пренебрежении членами порядка выше 2 дает формулу
![]() (8)
(8)
(∆R/R — относительная разность межатомных расстояний от замещающих друг друга ионов до их ближайшего окружения), константа в которой может быть выражена через молярный объем (V) и коэффициент сжимаемости (к).
И. Вазашерна и В. Хови получили для энтальпии смешения формулу
![]() (9)
(9)
, учитывая близость значений V/к для кристаллов сходного структурного типа, выражая эти величины через константу Маделунга (А) и потенциал межатомного отталкивания j(R): V/к = -[AzMzxe2/9R][Rj”(R)/j`(R) + 2] (zM, zx - зарядовые числа катиона и аниона соответственно; е — элементарный заряд), используя формулу
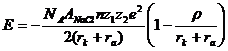 ,
,
получил уравнение для энергии решетки бинарных соединений MkXi
![]()
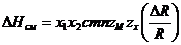 (10)
(10)
где z ‑ координационное число замещаемого иона; т = к + I; с ‑ эмпирический параметр, примерно постоянный для данного класса соединений (по структурному типу и зарядам атомов), имеющий размерность Дж/моль.
Для случая, когда химическая связь в твердом растворе не может считаться чисто ионной, B. C. Урусов предложил формулу
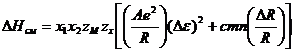 (11)
(11)
где ∆e — разность степеней ионности связи.
В случае образования твердых растворов d-элементов в формуле (11) должно присутствовать дополнительное слагаемое, отвечающее изменению энергии стабилизации кристаллическим полем. При этом расчетные и экспериментальные линии купола распада часто оказываются довольно близкими (рисунок 1).
|
Рисунок 1 – Линии купола распада некоторых бинарных систем: а) PbS-PbTe; б) CaS-MnS; в) CaS-MgS; г) VC-NbC (пунктирные линии – результаты расчета по выражению (11); сплошные – экспериментальные данные |
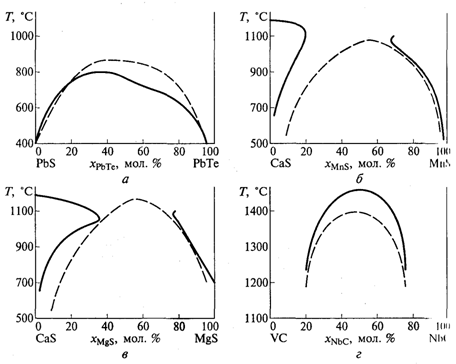
Представленный подход может быть использован не только для предсказания областей существования твердых растворов, но и для объяснения распределения катионов по кристаллографическим позициям в соединениях, различные компоненты которых могут занимать одни и те же позиции.
3. Энергия кристалла. Строгий теоретический расчет термодинамических свойств твердых соединений ‑ очень трудная задача. Трудности обусловлены в первую очередь достаточно сложным характером химической связи в реальных кристаллах. Поэтому широкое распространение получили различные приближенные и эмпирические вычислительные методы. Наиболее близки к неэмпирическим методы, основанные на описании модели межатомного взаимодействия как суперпозиции легко описываемых типов связи (например, чисто ионная, чисто металлическая или чисто ван-дер-ваальсовая связь) с подгонкой нескольких параметров модели к имеющимся экспериментальным данным (колебательным спектрам, модулям упругости, параметрам элементарной ячейки и т. д.) или расчете этих параметров для более простых соединений со сходным ближайшим координационным окружением атомов (например, при расчете свойств сложных оксидов параметры межатомного взаимодействия рассчитывают для простых оксидов). При этом в качестве стандартного состояния принимают разреженный газ не взаимодействующих атомов (или молекул в случае молекулярного кристалла).
Для упоминавшегося приближения чисто ионной связи энергию каждого иона кристаллической решетки обычно описывают формулой Борна –Майера‑Букингема

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 |
