

Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
![]() (12)
(12)
в которой параметры межатомного отталкивания (С, r) и ван-дер-ваальсового взаимодействия (В) подбирают эмпирически (z ‑ зарядовое число данного иона; R ‑ расстояние от данного иона до ближайших соседей; А ‑ константа Маделунга, которая включаем зарядовые числа ионов, взаимодействующих с рассматриваемым ионом).
Поскольку потенциал, задаваемый формулой (12), сферически симметричен, в случае ионов с существенной асимметрией координационного полиэдра (например, для ян-теллеровских ионов Мn3+ или Сu2+) вводят различные параметры взаимодействия в разных кристаллографических направлениях.
Металлический вклад в межатомные взаимодействия может быть оценен в рамках приближения ионного остова, взаимодействующего с электронным газом. Энергия каждого атома кристаллической решетки при этом составит
![]() (13)
(13)
Константа а в слагаемом, отвечающем за связывание атомов электронным газом, определяется кристаллической структурой вещества (включает константу Маделунга) и степенью ионизации атомного остова (а следовательно ‑ и концентрацией электронного газа). Константа b, отвечающая за энергию отталкивания за счет «давления» электронного газа, определяется кроме степени ионизации еще и эффективной массой электрона 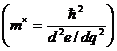 . В связи со сложностью определения этих параметров для реальных кристаллов константы а и b обычно подбирают эмпирически.
. В связи со сложностью определения этих параметров для реальных кристаллов константы а и b обычно подбирают эмпирически.
Для молекулярных кристаллов с ван-дер-ваальсовой связью между молекулами потенциал взаимодействия между атомами разных молекул задается потенциалом Леннард-Джонса
![]() (14)
(14)
или
![]() (15)
(15)
где А, В, С, r — эмпирически подбираемые константы; отметим, что параметр В может быть рассчитан и из поляризуемости, дипольного момента и потенциала ионизации.
Взаимодействие атомов одной молекулы часто (в случае значительно более высокой прочности внутримолекулярных связей по равнению с межмолекулярными) может быть удовлетворительно описано гармоническим потенциалом. При расчете (в отличие электростатического потенциала) может учитываться взаимодействие только между атомами нескольких ближайших координационных сфер, что связано с быстрым убыванием ван-дер-ваальсового потенциала притяжения с расстоянием (~R-6).
Применение эмпирических ангармонических межатомных потенциалов (например, широко используемого в спектроскопических исследованиях потенциала Морзе) с подбором параметров взаимодействия на основе экспериментальных данных для решения поставленной задачи не пригодно. Это связано с тем, что эксперимент дает информацию об атомах, колеблющихся вблизи положения равновесия, а для расчета энергии кристалла необходимо знать глубину потенциальной ямы по отношению к набору атомов (или молекул), находящихся на бесконечно большом расстоянии. Это приводит к необходимости использования только потенциалов, основанных на конкретной физической модели взаимодействия, и точность расчетов резко падает в случае значительного отклонения реального потенциала межатомного взаимодействия от модельного.
Подобные методы дают возможность получить «нулевую энергию» кристалла, входящую в уравнения как неопределенное слагаемое (Uo). Температурно-зависимая часть внутренней энергии при этом может быть вычислена по формулам
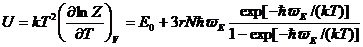 (16)
(16)
и
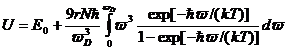 (17)
(17)
или в общем случае по формулам
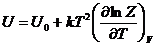 (18)
(18)
и
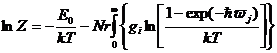 (19)
(19)
с использованием рассчитанного спектра плотности фононных состояний.
Расчет энтропии несколько проще, поскольку кристаллы как высокоупорядоченные системы характеризуются малыми значениями энтропии. При температуре 0 К энтропия кристаллов определяется степенью вырожденности низшего энергетического состояния (S = klng(Emin)) и при отсутствии вырождения равна нулю (постулат Планка). Температурно-зависимая часть энтропии может быть (как и в случае энтальпии) рассчитана по формулам
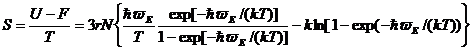 (20)
(20)
и
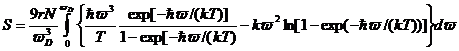 (21)
(21)
или в общем случае по уравнениям
![]() (22)
(22)
и (19).
Малые значения энтропии твердых тел позволяют пренебречь в первом приближении изменением энтропии в твердофазных процессах (правило Келли ‑ Кубашевского). Это правило не выполняется при участии в процессе (в качестве исходного вещества или продукта реакции) сильно дефектных соединений (например, сильно нестехиометрических оксидов Fe1-хO, ZnI+xO), сложных оксидов с значительным перераспределением катионов по кристаллографическим позициям (например, со структурой шпинели АВ2О4), суперионных проводников (например, RbAg4I5 и др.). В этом случае твердое тело часто может быть рассмотрено как раствор с энтропией смешения, определяемой формулой (2).
Подобные расчеты обеспечивают удовлетворительную точность не для всех кристаллических соединений. Поэтому был разработан ряд эмпирических методов, позволяющих рассчитывать изменения термодинамических функций (в первую очередь ‑ энтальпии и энтропии) твердофазных систем при протекании химических процессов по известным данным для ряда систем аналогичных веществ.
4. Анализ энергетики координационного окружения. на основании анализа большого числа термохимических и кристаллохимических данных для различных простых и сложных оксидов в разных полиморфных модификациях пришел к выводу, что энтальпии твердофазных процессов могут линейно зависеть от энергий предпочтения тем или иным катионом того или иного координационного полиэдра:
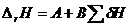 (23)
(23)
где А, В — константы для конкретного типа реакций веществ данной природы; SdН — сумма энергий изменения координационного окружения (главным образом ‑ координационных чисел) катионов, которые в свою очередь могут рассматриваться как индивидуальные характеристики того или иного катиона.
Расчет по формуле (23) дает значения энтальпий химических реакций с довольно большой погрешностью, однако в ряде случаев может быть полезен для получения оценочных данных.
Метод сравнительного расчета Карапетьянца. Сопоставляя значения ∆Н или ∆S для двух типов реакций с участием одного и того же ряда элементов (например, лантаноидов), можно вычислить неизвестные для какого-либо элемента термодинамические характеристики одной из реакций по линейному соотношению {А, В ‑ параметры):
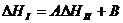 ;
; 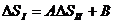 (24)
(24)
Например, сравнение энтальпий образования из простых оксидов вольфраматов состава Ln2O3-WO3 и 3Ln2O3-WO3 (3Gd2O3WO3 и 3Nd2O3-WO3) позволило определить неизвестные теплоты образования большого числа вольфраматов 3Ln2O3- WO3.
Удовлетворительные результаты данный метод дает при исследовании термодинамических характеристик сульфидов, селенидов и теллуридов. Сходное выражение может быть записано и для энтропии двух рядов соединений:
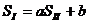 (25)
(25)
Которое также было с успехом использовано для расчета энтропии большого числа сульфидов, селенидов и теллуридов.
Корреляция энтальпии (энтропии) со структурными характеристикам. Для рядов соединений со сходной структурой можно ожидать корреляции между структурными и термодинамическими параметрами. Простейшим случаем, который часто представляется и наиболее целесообразным с учетом погрешностей расчета, является линейная зависимость
∆Н = аР + b; ∆S = аР + b, (26)
где Р — параметр, связанный со структурой кристалла.
Например, для многих рядов сложных оксидов или халькогенидов АВХ3 с перовскитоподобной структурой уравнение (26) хорошо выполняется как для энтропии соединений, так и для изменения энтальпии процессов с их участием. В уравнении (26) параметр Р ‑ фактор толерантности Гольдшмидта:
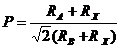
где RA, RB, Rx — ионные радиусы соответствующих ионов.
В качестве параметра Р могут выступать также степень искажения решетки с понижением симметрии, молярный объем (V), плотность (r) и т. д. В частности, линейная корреляция между энтропией и величиной p-l/2V-l для оксидов со структурой рутила GeO2, SnO2, PbO2 позволила с хорошей точностью предсказать энтропию высокоплотной модификации SiO2 (стишовита).
5. Структура веществ ‑ участников реакции. При сравнении структуры MgAl2O4 со структурами MgO и А12О3 выявляются одновременно признаки сходства и различия. И MgO, и шпинель имеют плотнейшую кубическую упаковку ионов кислорода, тогда как Аl2О3 ‑ искаженную гексагональную плотнейшую упаковку. В то же время ионы А13+ занимают как в А12О3, так и в шпинели октаэдрические позиции, а ионы Mg2+ ‑ октаэдрические позиции в MgO, но тетраэдрические в MgAl2O4.
6. Закономерности зародышеобразования в твердофазных системах. Гетерогенные фазовые превращения протекают через образование и последующий рост зародышей второй фазы (или нескольких фаз). При этом описание термодинамики систем, включающих малые частицы (какими являются зародыши на начальных стадиях своего роста), должно учитывать свойства поверхности раздела фаз. Увеличение площади поверхности раздела фаз приводит к прямо пропорциональному росту части энергии Гиббса, откуда следует неравновесность избыточной поверхности раздела, наблюдающейся в дисперсных системах.

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 |
