

Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Публикация доступна для обсуждения в интернет как материал “Всероссийской рабочей
химической конференции “Бутлеровское наследие-2011”. http:///bh-2011/
Поступила в редакцию 20 апреля 2011 г. УДК 547:544.424.
Изучение межмолекулярных комплексов диметилселенида
с v-акцепторами: квантовохимические аспекты
© ,1+ 1*
и Angel Martín Pendás2
1 Кафедра органической химии. Химический институт им. . Казанский (Приволжский) федеральный университет. . г. Казань, 420008. Республика Татарстан. Россия. Тел.: (843) 233-77-89. E-mail: Timur.Madzhidov@ksu.ru, Galina.Tschmutowa@ksu.ru
2 Departamento de Quimica Fisica y Analitica.Facultad de Quimica.
Universidad de Oviedo, Avda. Julián Clavería, 8. Oviedo, 33006. Spain.
E-mail: *****@***chimica. unovi. es
_______________________________________________
*Ведущий направление; +Поддерживающий переписку
Ключевые слова: селеноорганические соединения, межмолекулярные комплексы, v-акцепторы, теория атомов в молекулах, подход взаимодействующего квантового атома, природа межмолекулярных взаимодействий.
Аннотация
С использованием метода теории функционала плотности (B3LYP/6-311++G(d, p)) исследованы комплексы диметилселенида с несколькими v-электроноакцепторами разной силы. Обнаружено, что обобществление электронной плотности играет более важную роль, нежели электростатическое взаи-модействие между компонентами комплексов. С использованием анализа топологии распределения электронной плотности выявлено, что в зависимости от характера электроноакцептора природа связи между компонентами меняется от ван-дер-ваальсовой до слабой ковалентной. С использованием подхода взаимодействующего квантового атома на примере комплекса H2Se…AlH3 были проана-лизированы вклады от электростатических и обменно-корреляционных взаимодействий в связь Se…Al и межмолекулярное связывание в целом.
Введение
Химия селена в настоящее время является динамично развивающейся областью совре-менной химической науки [1, 2]. Помимо использования селеноорганических соединений в синтезе в качестве реагентов и катализаторов, в технологии для получения органических проводников и полупроводников [1, 3], а также квантовых точек с помощью селеноорга-нических соединений [4, 5]; все большее значение они приобретают в медицине для создания лекарственных препаратов и биологически активных добавок. Показана фундаментальная важность селена для здоровья живых организмов и человека в том числе [6]. Биохимическая роль селена в организме млекопитающих стала отчасти понятной после открытия участия селена в активном центре фермента глутатионпероксидазы [7, 8], однако число обнаружен-ных природных белков и ферментов, содержащих селен, в последнее время существенно возросло: селен входит в состав форматдегидрогеназ и гидрогеназ, глицинредуктаз, иодотиро-ниндеионидаз, тиоредоксинредуктаз, селенофосфатсинтетаз, различных селенобелков [6]. Отчасти и по этой причине в 1999 году селеноцистеин был признан IUPAC 21ой «природной» аминокислотой [9]. Атом селена является активным электронодонором, способным выступать в качестве слабого акцептора протона [10], достаточно сильного акцептора молекулы иода [11, 12], а также других льюисовых кислот [13, 14]. При этом атом селена как электронодонор проявляет достаточно необычные свойства, обусловливающие нетрадиционную природу комплексов с его участием.
В данной работе мы уделили внимание комплексам селеноорганических соединений с льюисовыми кислотами, называемыми v-акцепторами. С точки зрения теории валентных связей, комплексы с v-акцепторами характеризуются переносом электронной плотности с занятых орбиталей (НЭП) электронодонора на вакантные орбитали внешнего квантового слоя атома-электроноакцептора. Центры комплексообразования v-акцепторов чаще всего поло-жительно заряжены, и этот эффект, вкупе с положительным зарядом на атоме селена, отме-ченным ранее в работах [10, 12], может играть важную роль в межмолекулярном взаимо-действии. С другой стороны, наличие вакантных низко расположенных электронных оболо-чек способствует эффективному переносу заряда на них. Были исследованы комплексы диметилселенида как простейшего селеноорганического соединения с соединениями метал-лов и неметаллов различных типов, для которых имеются термохимические данные: AlBr3 (у комплекса с дипропилселенидом ΔHcomp298 = -29.9 ккал/моль [15]), BBr3 (у комплекса с дипро-пилселенидом ΔHcomp298 = -22.8 ккал/моль [16]), Zn(CH3)2 (энтальпия комплексообразования с диметилселенидом ΔHcomp298 = -1.5±0.5 ккал/моль [17]). Для выявления влияния окружения атома металла рассчитаны также комплексы с трифторидом бора.
Экспериментальная часть
Структуры всех исследуемых в работе соединений и комплексов получены с использованием программного пакета Gaussian03 [18] в результате полной оптимизации геометрии в газовой фазе. Все обсуждаемые структуры соответствуют минимуму поверхности потенциальной энергии, что прове-рялось расчетом гессиана, все члены которого являются действительными числами. Поиск глобаль-ного минимума производился с помощью использования разных стартовых геометрий. Расчеты комплексов проводились методом теории функционала плотности с использованием гибридного функционала B3LYP [19-21] в базисе 6-311++G(d, p). В расчетах были использованы жесткие критерии самосогласования поля, оптимизации геометрии и расчетов интегралов. Релятивистские поправки не вводились.
Ошибка суперпозиции базисного набора принималась во внимание a posteriori с использованием метода уравновешивающей функции Бойса-Бернарди [22] (далее обозначаемого как СР – от англ. counterpoise). Для оптимизации геометрии с учетом BSSE с помощью процедуры СР использовался подход Симона-Дюрана-Данненберга [23].
Топологические и интегральные характеристики атомов в рамках теории атомов в молекулах (АвМ) вычислялись с использованием программы AIMAll [24]. Требуемая для расчетов волновая функция получалась в том же методе, в каком проводились оптимизация геометрии и расчет частот. Приведенные в работе рисунки сгенерированы с использованием программы AIMStudio пакета AIMAll. Обсуждаемые величины зарядов получены интегрированием электронной плотности по бассейну атомов в рамках АвМ. NBO анализ [25] проведен c использованием стандартной процедуры, реализованной в программе Gaussian03.
Численное интегрирование по бассейну атомов в рамках подхода IQA (IQF) проведено с использованием программы Promolden [26]. В ходе интегрирования вводились бета-сфер на всех атомах такого размера, что их радиус равен примерно 80-90% расстояния до ближайшей критической точки. Внутри бета-сфер использованы радиальные квадратурные формулы Гаусса-Чебышева второго рода с 1500 узлами разбиения и угловые квадратурные формулы Лебедева с 1730 узлами; мультиполь-ное разложение основных интегралов было ограничено L≤6. Вне бета-сфер использовались квадратур-ные формулы трапеций с 1500 узлами разбиения в качестве радиальных и угловые квадратурные формулы Лебедева с 5810 узлами; мультипольное разложение обрывалось при L=10. Волновые функции, требуемые для проведения расчетов IQA (IQF), были получены методом Хартри-Фока без учета суперпозиционной ошибки при оптимизации геометрии и при расчете энергии взаимодействия. Для ускорения расчетов внутренние (остовные) орбитали в волновой функции моделировались с использованием псевдопотенциалов Хэя-Водта [27]. Валентные оболочки при этом моделировались базисом 6-311++G(3d,3p). Использованная методика расчета описана в работе [28]. Описание подхода взаимодействующего квантового атома и фрагмента приведено в приложении.
Теоретические основы подхода взаимодействующего квантового атома. Разработанный в работе [29] подход для экономного вычисления электрон-электронного отталкивания позволяет пред-ставить энергию молекулы как сумму собственной энергии атомов EselfA и энергии взаимодействия VintAB всех атомов системы: 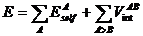 , где суммирование ведется по атомам и неиден-тичным парам атомов системы соответственно [26]. Собственная энергия атома представляет собой сумму кинетической энергии электронов, энергии притяжения электронов к ядру и межэлектронного отталкивания в пределах бассейна атома: EselfA = TA + VenAA + VeeAA. Энергия взаимодействия между атомами А и В есть сумма энергии ядерно-ядерного отталкивания, притяжения электронов атома А к ядру атома В и наоборот, а также отталкивания между электронами двух бассейнов: VintAB = VnnAB + VenAB + VneAB + VeeAB. Разбиение члена VeeAВ на кулоновское отталкивание VCAB и обменно-корреляционное притяжение между электронами VХCAB позволяет выделить член, описывающий только электростатическое (кулоновское) взаимодействие между атомами EelАВ = VnnAB + VenAB + VneAB + VCAB; оставшийся член (VХCAB) энергии межатомного взаимодействия представляет собой энергию обменно-корреляционного взаимодействия между атомами. Эта величина имеет чисто квантовую природу и характеризует ковалентность связывания. Изменение энергии атома при образовании связи называется энергией деформации атома EdefA. В таком случае энергия связывания есть сумма электростатического, обменно-корреляционного взаимодействия и энергии деформации атомов. В этом заключается подход взаимодействующего квантового атома (IQA).
, где суммирование ведется по атомам и неиден-тичным парам атомов системы соответственно [26]. Собственная энергия атома представляет собой сумму кинетической энергии электронов, энергии притяжения электронов к ядру и межэлектронного отталкивания в пределах бассейна атома: EselfA = TA + VenAA + VeeAA. Энергия взаимодействия между атомами А и В есть сумма энергии ядерно-ядерного отталкивания, притяжения электронов атома А к ядру атома В и наоборот, а также отталкивания между электронами двух бассейнов: VintAB = VnnAB + VenAB + VneAB + VeeAB. Разбиение члена VeeAВ на кулоновское отталкивание VCAB и обменно-корреляционное притяжение между электронами VХCAB позволяет выделить член, описывающий только электростатическое (кулоновское) взаимодействие между атомами EelАВ = VnnAB + VenAB + VneAB + VCAB; оставшийся член (VХCAB) энергии межатомного взаимодействия представляет собой энергию обменно-корреляционного взаимодействия между атомами. Эта величина имеет чисто квантовую природу и характеризует ковалентность связывания. Изменение энергии атома при образовании связи называется энергией деформации атома EdefA. В таком случае энергия связывания есть сумма электростатического, обменно-корреляционного взаимодействия и энергии деформации атомов. В этом заключается подход взаимодействующего квантового атома (IQA).
Распространение данного подхода на несколько взаимодействующих многоатомных фрагментов носит название подхода взаимодействующего квантового фрагмента (IQF) [30]. При образовании связи между двумя фрагментами, помимо образования новых атом-атомных взаимодействий (энергия которых находится по тем же формулам, что и выше), происходит деформация электронной и прост-ранственной структуры фрагментов, а также изменение прочности связей внутри них. Величина энер-гии деформации фрагмента Х, Edef, оценивается как сумма между изменениями собственных энергий атомов фрагмента EdefA = ΔTA + ΔVenAA + ΔVCAA + ΔVXCAA и изменением энергии взаимодействия между соответствующими атомами при комплексообразовании ΔVintAB = VintAB(комплекс) - VintAB (изолированный фрагмент): ![]() , где суммирование ведется по всем атомам и неиден-тичным парам атомов фрагмента Х соответственно. В таком случае энергия взаимодействия между атомами есть:
, где суммирование ведется по всем атомам и неиден-тичным парам атомов фрагмента Х соответственно. В таком случае энергия взаимодействия между атомами есть: ![]() , где первое суммирование ведется по всем фрагментам, а второе и третье – по всем возможным неидентичным парам атомов различных фрагментов. В случае отсутствия ошибки интегрирования данная величина в точности должна равняться разнице в электронных энергиях комплекса и составляющих его компонентов. Первый и последний члены при объединении дают обменно-репульсионный член XRC, сходный с таковым в некоторых других методах разбиения энергии взаимодействия. Поскольку в методе Хартри-Фока электронная корреляция не учитывается, обменно-корреляционный член в нем вырождается в чисто обменный ΔVXAA.
, где первое суммирование ведется по всем фрагментам, а второе и третье – по всем возможным неидентичным парам атомов различных фрагментов. В случае отсутствия ошибки интегрирования данная величина в точности должна равняться разнице в электронных энергиях комплекса и составляющих его компонентов. Первый и последний члены при объединении дают обменно-репульсионный член XRC, сходный с таковым в некоторых других методах разбиения энергии взаимодействия. Поскольку в методе Хартри-Фока электронная корреляция не учитывается, обменно-корреляционный член в нем вырождается в чисто обменный ΔVXAA.

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 |
