
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Установленные отклонения времен τd для системы «гидразин – сжиженный кислород» при нагревании металлическими (сталь, алюминий) и неметаллическими (керамика, углерод) частицами малых размеров (рис. 2) показывают, что с понижением температуры локального источника энергии влияние его теплосодержания усиливается. При равных размерах и начальных температурах теплосодержание стальных, алюминиевых, керамических и углеродистых частиц существенно отличается. Теплосодержание частиц можно вычислить по следующему выражению [25]:
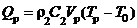 ,
,
где Vp – объем частицы, м3 (![]() ).
).
При Θp=1.1 и Rd=Zd=0.15: Qp=0.315 кДж – сталь; Qp=0.208 кДж – алюминий; Qp=0.302 кДж – керамика; Qp=0.161 кДж – углерод.
Рис. 2 показывает, что имеется некоторое предельное значение Qp, при превышении которого материал источника нагрева из рассматриваемых комбинаций несущественно влияет на инерционность зажигания. Так, например, можно выделить значение Qp≈0.35 кДж при Θp=1.2.
Максимальным значением теплоты Qp, аккумулированной стальной частицей, можно объяснить минимальные времена задержки зажигания топлива τd (рис. 2). Наибольшие времена τd характерны углеродистой частице с минимальным значением Qp. Несмотря на большее теплосодержание керамической частицы по сравнению с алюминиевой времена задержки зажигания для последней меньше. Это можно объяснить существенным превышением теплопроводности алюминия по сравнению с керамическими материалами (алюминий – λ=98 Вт/(мК), керамика – λ=20 Вт/(мК)), а также поступлением в зону реакции дополнительной энергии в результате экзотермического перехода при кристаллизации расплава металла. Приповерхностный слой топлива прогревается быстрее при нагревании алюминиевой частицей, которая вследствие больших значений λ интенсивно остывает. Температура керамической частицы изменяется несущественно (в пределах 3 %) относительно начальной Θp даже при относительно больших временах задержки зажигания (τd≈0.2). Температура алюминиевой частицы на границе контакта с топливом при τd≈0.2 уменьшается относительно начальной на 7 %.
Важно отметить, что отклонения времен τd для жидких топлив при зажигании их стальными, алюминиевыми и углеродистыми частицами [31] существенно больше (до 43 %) полученных (рис. 2) для рассматриваемой системы (до 18 %). Это можно объяснить тем, что характерные времена τd, приведенные в [31], имеют значения, в несколько раз превышающие времена задержки зажигания, представленные на рис. 2. При возрастании времен процесса зажигания усиливается влияние фактора остывания локального источника энергии [31]. Как следствие определяющую роль играют его теплофизические характеристики.
Известно [4–10], что при локальном нагреве твердых и жидких топлив помимо температуры источника существенное влияние играют и его размеры. Для рассматриваемой гелеобразной системы (рис. 1) установлено, что варьирование размеров металлических и неметаллических частиц Rp и Zp в достаточно широком диапазоне (0.05<Rp<0.25, 0.05<Zp<0.25) не приводит к существенному изменению τd. Можно выделить некоторые минимальные размеры частицы – Rp=Zp=0.075, при превышении которых времена τd практически не изменяются (менее 5 %). В условиях незначительного влияния размеров Rp и Zp (при Rp=Zp>0.075) определяющую роль играют зависимости τd=f(Θp). В тоже время важно отметить, что влияние размеров на инерционность зажигания усиливается при значениях Θp, соответствующих нижним границам диапазонов изменения температуры источника нагрева, при которых возможно зажигание (нижним пороговым значениям Θp).
Показано [4–10], что условия зажигания твердых и жидких конденсированных веществ могут достаточно существенно отличаться при изменении теплосодержания одиночных разогретых металлических и неметаллических частиц. В частности, возможна реализация нескольких режимов зажигания, отличающихся как положением зоны ведущей реакции окисления относительно границы контакта источника нагрева и топлива, так и предельными значениями Θp, Rp и Zp. Для рассматриваемой гелеобразной топливной композиции установлено, что при варьировании основных параметров металлических и неметаллических частиц в широких диапазонах (1.1<Θp<1.5, 0.05<Rp<0.25, 0.05<Zp<0.25) изменения режима зажигания не происходит – зона ведущей реакции окисления формируется в малой окрестности поверхности топлива и боковой грани частицы.
На рис. 3, а показаны изотермы в момент воспламенения при Θp=1.1 и Rp=Zp=0.15. Следует отметить, что инертный газ, заполняющий в начальный момент времени область над частицей, прогревается существенно быстрее, чем поступающие с поверхности топлива (из зоны эндотермических фазовых переходов) компоненты горючего и окислителя. Между зонами интенсивной реакции окисления (Z→Z1) и прогретого газа (Z→Z2) имеются участки (Z1<Z<Z2) пониженной температуры (рис. 3, а). Формирование таких областей можно объяснить тем, что при остывании частицы снижается интенсивность прогрева смеси инертного газа с продуктами парообразования. Рост температуры парогазовой смеси вблизи боковой грани частицы при τ→τd (рис. 3, а) можно объяснить тепловыделением при ускорении реакции окисления. При временах τ>τd направления тепловых потоков меняются на противоположные – несколько остывшая частица нагревается за счет тепла воспламенившейся парогазовой смеси.
а
б
в
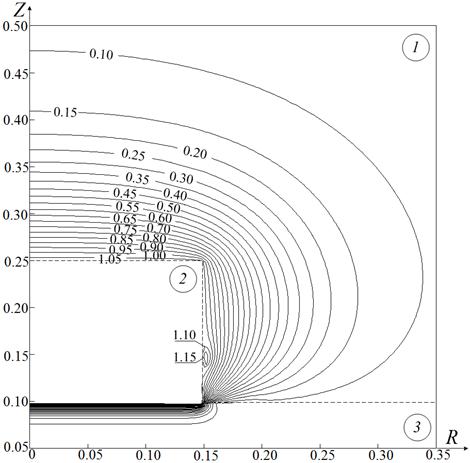
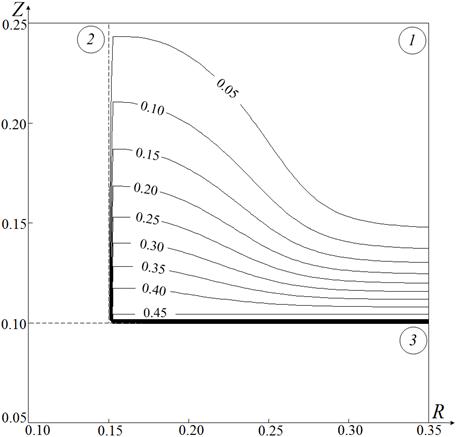
Рис. 3. Изотермы Θ (а), изолинии концентраций горючего Cf (б) и окислителя Co (в) для системы «гидразин – сжиженный кислород – керамическая частица»
в момент воспламенения при τd=0.18, Θp=1.1, Rp=Zp=0.15, Zc=0.1
На рис. 3, б, в показаны изолинии концентраций горючего и окислителя в момент воспламенения при Θp=1.1 и Rp=Zp=0.15. Видно, что концентрация окислителя Co в окрестности поверхности топлива растет в несколько раз быстрее, чем концентрация горючего Cf. Это обусловлено значительным отличием скоростей испарения гидразина и сжиженного кислорода [11, 12]. Как следствие реакция окисления ускоряется при высоких значениях Co и умеренных значениях Cf.
Установленную особенность реализации одного режима зажигания типичной гелеобразной топливной композиции – «гидразин – сжиженный кислород», характеризующегося относительно небольшими концентрациями горючего Cf и окислителя Co (рис. 3, б, в), достаточными для воспламенения, малыми временами задержки зажигания τd (рис. 2) по сравнению с типичными твердыми и жидкими топливами [4–10], специфическим расположением зоны ведущей реакции окисления относительно границы контакта источника нагрева и топлива (рис. 3, а), можно объяснить с использованием основных положений современной теории зажигания конденсированных веществ при локальном нагреве [31].
Время задержки зажигания топлива τd можно условно [31] представить в виде суммы двух слагаемых (физическая и химическая составляющие). Физическая составляющая τd – время прогрева приповерхностного слоя топлива, фазовых переходов и прогрева газообразных компонентов. Химическая составляющая τd – время, в течение которого происходит интенсивная химическая реакция окисления паров горючего, принимающая взрывной характер (ускорение реакции окисления с резко экспоненциальным увеличением выделяемой энергии). Выделение таких составляющих τd условное, так как физико-химические процессы протекают одновременно. Однако по такому принципу можно определить наиболее инерционные стадии процесса зажигания.
Для вычисления химической составляющей τd целесообразно [31] использовать классическое выражение для времени индукции [21]:
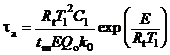 .
.
Кинетические параметры реакции окисления (Qо, E, k0) газообразных компонентов рассматриваемой гелеобразной структуры [26, 27] соответствуют большим скоростям Wo по сравнению со скоростями химического реагирования в системах с твердыми и жидкими конденсированными веществами [4–10]. Поэтому времена τa существенно меньше аналогичных параметров для жидких и твердых топлив, рассмотренных в [4–10].
Время прогрева приповерхностного слоя вещества и компонентов парогазовой смеси определялись по формуле [31]:
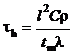 ,
,
где l – характерный размер прогретого слоя вещества, м.
Результаты выполненных численных исследований показали, что для рассматриваемой гелеобразной топливной композиции характерный размер прогретого приповерхностного слоя вещества (рис. 3, а) меньше описанных в [4–10, 31]. Теплофизические характеристики взаимодействующих веществ (λ, С, ρ) сопоставимы с используемыми при моделировании в [4–10, 31]. Поэтому времена τh для рассматриваемой гелеобразной топливной композиции также меньше времен τh, приведенных в [31].
Анализ характерных времен τd показывает, что для реализации условий воспламенения необходимо подвести количество энергии, достаточное для плавления приповерхностного слоя топлива и последующего испарения. Установлено, что при Θp>1.2 теплосодержания металлических и неметаллических частиц различной физической природы достаточно для ускорения реакции окисления при относительно небольших временах τh и τa. При уменьшении Θp до 1.1 возрастают значения составляющих τh и τa.
Важно отметить более интенсивное по сравнению с твердыми и жидкими топливами остывание частиц при контакте с гелеобразным топливом в течение времени τd. Это также объясняется энергозатратными эндотермическими фазовыми переходами и криогенными температурами хранения топлива. Установленная особенность процесса позволяет сделать вывод о том, что если реакция окисления не ускорилась при относительно небольших временах τd<<1, то вероятность зажигания уменьшается, так как и металлические и неметаллические частицы интенсивно остывают при τd→1.

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 |
