

Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Существенное изменение энтальпий образования в орто-изомерах соединений с водородсодержащими заместителями может быть связано с образованием внутримолекулярных водородных связей. Так, например, энтальпия образования о-нитрофенола на 30.0 и 22.2 кДж/моль ниже энтальпий образования м - и п-нитрофенолов соответственно. Для о-нитроанилина эти различия заметно меньше - 12.2 и 2.5 кДж/моль соответственно. Эти данные, а также анализ основных геометрических параметров и барьеров вращения изомерных нитрофенолов и нитроанилинов указывает на наличие сильной внутримолекулярной водородной связи в о-нитрофеноле и существенно более слабой в о-нитроанилине. С этим выводом согласуются и имеющиеся экспериментальные, прежде всего спектральные [24] данные.
2 Влияние строения молекул на энергию активации радикального газофазного распада нитроаренов 2.1 Изучение взаимосвязи изменения энергии диссоциации связи C-N и параметров геометрической и электронной структуры нитроареновПроцесс газофазного распада ароматических нитросоединений протекает достаточно сложно, поэтому экспериментальное определение кинетических параметров мономолекулярного распада представляет собой очень трудную задачу. Отсутствие надежных теоретических оценок энергии различных альтернативных вариантов первичного акта, как уже отмечалось выше, не позволяет делать каких-либо однозначных выводов о механизме процесса на основе анализа только экспериментальных данных. Использование квантово-химических оценок энергии диссоциации связи C-NO2 существенно меняет ситуацию.
Величины D(C-N) в изученных молекулах рассчитывались на основе разности энтальпий образования радикалов и исходного соединения Полученные результаты представлены в таблице 5.
Расчетные энергии диссоциации связи C-N в целом близки к термохимическим оценкам для нитробензола. За исключением нитротолуолов, для которых можно ожидать большую ошибку в определении энергии активации газофазного распада, различие квантово-химических значений D(C-N) и экспериментально измеренных величин энергии активации, не превышает 10 – 13 кДж/моль, что сопоставимо с приводимой погрешностью экспериментального определения. Выше мы отмечали значительное различие экспериментальных и расчетных значений энтальпий образования ароматических нитросоединений и радикалов. Подобное достаточно хорошее согласие в данном случае может быть связано с тем, что значительные ошибки в энтальпиях образования ароматических нитросоединений при вычислении D(C-N) частично компенсируются за счет погрешности определения энтальпий образования соответствующих радикалов.
Таблица 5 Энергии диссоциации (кДж/моль) связи C-N ароматических нитросоединений по данным метода B3LYP/6-31G(d). В скобках приведены данные метода MP2/6-31G(d) [27].
Соединение | D(C-N) | Соединение | D(C-N) |
нитробензол | 292.6 (290.6) | 1,3,5-тринитробензол | 271.0 |
о-нитротолуол | 281.9 (281.7) | 2,4,6-тринитротолуол | 249.7 |
м-нитротолуол | 293.3 (293.3) | 2,4,6-тринитротолуол | 275.8 |
п-нитротолуол | 296.8 (293.4) | о-фторнитробензол | 275.0 |
о-нитроанилин | 310.6 (302.8) | м-фторнитробензол | 287.3 |
м-нитроанилин | 294.6 (298.3) | п-фторнитробензол | 295.0 |
п-нитроанилин | 310.5 (304.1) | о-хлорнитробензол | 261.3 |
о-нитрофенол | 326.1 (301.3) | м-хлорнитробензол | 285.2 |
м-нитрофенол | 290.2 (290.4) | п-хлорнитробензол | 291.2 |
п-нитрофенол | 303.1 (294.6) | о-бромнитробензол | 263.4 |
о-динитробензол | 244.8 | м-бромнитробензол | 285.1 |
м-динитробензол | 280.8 | п-бромнитробензол | 290.8 |
п-динитробензол | 279.7 |
Полученные результаты показывают, что донорные заместители повышают энергию диссоциации, а акцепторные заместителями (в нашем случае только NO2-группа) ее снижают. Наиболее сильно эффект увеличения прочности связи C-N наблюдается для всех пара-замещенных. Это объясняется с позиций проявления прямого полярного сопряжения донорного заместителя с NO2-группой. Для мета-замещенных прямое полярное сопряжение невозможно, поэтому наблюдается меньшее значение энергии диссоциации по сравнению с пара-изомерами. Сопоставление результатов таблиц 5 и 1 показывает, что в изменении r(C-N) и D(C-N) существует определенная связь, наиболее наглядно она проявляется для п-замещенных нитробензола (рисунок 2). Соответствующая корреляционная зависимость может быть использована для оценки D(C-N) нитробензолов, если имеются сведения о геометрии молекулы.
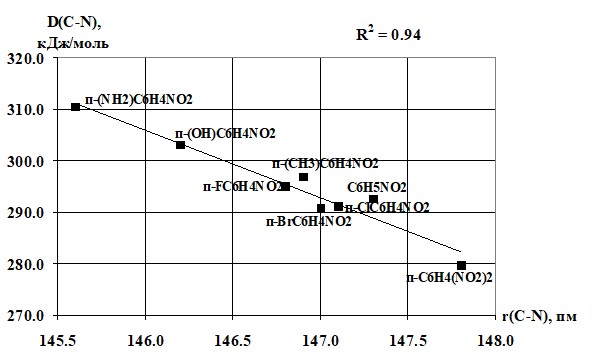
Рисунок 2 Зависимость энергии диссоциации связи C-N от длины этой связи (коэффициент корреляции 0.94).
Отметим, что достаточно близкие значения энергий диссоциации связи C-NO2 в ряду монофункциональных производных нитробензола были получены и с использованием метода MP2/6-31G(d) [28]. В этом случае значения D(C-N) рассчитывались не из энтальпий образования соединения и радикалов, а из полных электронных энергий. Наличие независимых хорошо согласующихся между собой квантово-химических данных, полученных с использованием различных методов, может служить важным дополнительным аргументом в пользу радикального механизма первичного акта газофазного мономолекулярного распада изученных ароматических нитросоединений.
Экспериментальные значения энергии активации первичного акта и и расчетные значения энергии диссоциации связи C-NO2 достаточно близки. Для большинства изученных соединений согласованно изменяются и тенденции изменения в ряду. Исключение составляют мета - и пара-нитротолуолы, для которых экспериментальные и расчетные значения энергии (энтальпии) активации существенно различаются. Согласно расчетным оценкам величины D(C-N) для м-нитротолуола практически совпадает с соответствующими значениями для нитробензола. Для п-нитротолуола расчет предсказывает небольшое увеличение D(C-N) (на 4.2 кДж/моль), а эксперимент – существенное уменьшении энергии активации (более чем на 10 кДж/моль) по сравнению с нитробензолом. Отметим, что анализ тенденций изменения параметров геометрической и электронной структуры в ряду ароматических нитросоединений согласованно указывает на повышение прочности связи C-NO2 в нитротолуолах. В работе [27], в которой этот вопрос впервые подробно анализировался, был сделан вывод о том, что экспериментальные значения энергии активации термического распада м-нитротолуола и п-нитротолуола занижены, что может быть связано с наличием в указанных соединениях примесей о-нитротолуола или с вкладом в эффективную константу скорости более быстрых вторичных реакций. Полученные нами с использованием метода B3LYP/6-31G(d) результаты подтверждают этот вывод. Наряду с отмеченными в [27] причинами, энергия активации термического распада мета - и пара-нитротолуолов может отличаться от D(C-N) и за счет вклада альтернативных механизмов нерадикального распада с меньшим значением энергии активации, например параллельным протеканием ННП.
Другой, необходимой для расчета константы скорости реакции мономолекулярного распада величиной является предэкспоненциальный множитель реакции. Чтобы строго рассчитать предэкспоненциальный множитель необходимо иметь сведения о структуре ПС и соответствующих статистических суммах ПС и исходной молекулы. Для реакции радикального распада это сложная задача. Но получить приближенные оценки о величине предэкспоненциального множителя можно и существенно более простым путем.
Как уже отмечалось, строгий теоретический расчет предэкспоненциального множителя мономолекулярного распада предполагает определение структуры активированного комплекса и оценку составляющих энтропии активации, отвечающих различным видам движения: вращательному, колебательному и внутреннему вращению. Для реакции радикального распада наиболее сложной проблемой является оценка критического расстояния рвущейся связи, отвечающей активированному комплексу.
Мы уже приводили данные, что для реакций нитрометана, нитроэтана и нитроэтилена на поверхности потенциальной энергии реакции радикального распада вдоль координаты реакции отсутствуют характеристические точки (очевидно, аналогичная ситуация наблюдается и для ароматических нитросоединений; к сожалению, из-за ряда технических трудностей нам не удалось завершить подобный расчет для нитробензола), что еще больше осложняет расчет предэкспоненциального множителя. Поэтому в данном разделе мы приводим только результаты оценки предэкспоненциального множителя с использованием ряда упрощающих предположений. При этом мы не ставили задачу строго рассчитать k0, а только пытались выяснить влияние на его величину барьера вращения нитрогруппы.
Существует формула, связывающая величину предэкспоненциального множителя мономолекулярного радикального распада с энтропией активации:
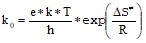 (1)
(1)
где e = 2.718;
k = 1.381*10-23 Дж/K – постоянная Больцмана;
T = 298.15 K – температура;
h = 6.626*10-34 Дж*с – постоянная Планка;
ΔS≠ - энтропия активации;
R = 8.315 Дж/(K*моль) – молярная газовая постоянная.
Из формулы 1 следует, что при постоянной температуре предэкспоненциальный множитель определяется величиной энтропии активации. В свою очередь, энтропия активации есть, как известно, разность значений энтропий ПС и исходной молекулы. Но оказывается, и это подтверждается и анализом литературы и проведенными расчетами, что в ряду соединений близкого строения величина энтропии активации реакции радикального распада нитросоединений определяется, в основном, значением статистической суммы внутреннего вращения исходных молекул. Из данных, приведенных в таблице 6 следует, что величина энтропии реакции для большинства монофункциональных производных нитробензола изменяется незначительно, поэтому вклад в энтропию активации вносит, в основном, энтропия внутреннего вращения.

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 |
