

Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
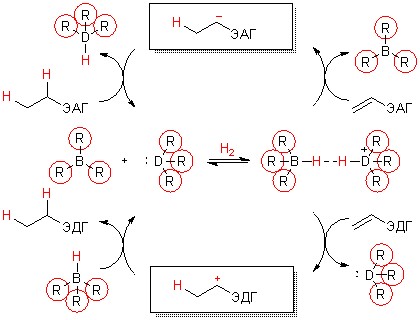
Схема 1. Механизмы гидрирования, катализируемого РПЛ, различных двойных связей.
ЭАГ – электроноакцепторная группа, ЭДГ – электронодонорная группа.
Таким образом, целью данной работы стало изучение механизма реакций, относящихся ко второй группе. Для этого было решено провести кинетические измерения количественной реакционной способности (нуклеофильности) следующих борогидридов: HB(C6F5)3-, HB(2,4,6-F3C6H2)3- и HB(2,6-F2C6H3)3-, которые являются гидридными переносчиками в гидрировании алкенов с электроноакцепторными группами (ЭАГ), катализируемым РПЛ[3,4]. Для измерения нуклеофильности мы выбрали «бензгидрильную» методологию, разработанную ранее в нашей лаборатории[5].
Борогидриды HB(Ar)3- с различными противоионами были синтезированы по реакции эквимолярных количеств триарилборанов, аммоний - или фосфониевых галогенидов и триэтилсилана в дихлорметане при 20 °C. Данный метод мы примени ли для синтеза триалкиламмониевых (R3NH+) и триалкилфосфониевых (R3PH+) борогидридов, которые ранее было возможно синтезировать только по реакции водорода со смесью соответствующих кислот и оснований Льюиса при повышенном давлении.
Кинетика реакций бензгидрильных ионов с борогидридами были изучены в условиях псевдо-первого порядка ([R3BH-] >> [Ar2CH+]) с помощью УФ или видимой спектроскопии при ?max поглощения электрофилов. Обработка моно-экспоненциальных кривых спада поглощения бензгидрильных катионов позволила получить значения констант скорости второго порядка (k2) для реакций борогидридов с бензгидрильными катионами. Зависимости log(k2) от параметров электрофильности E соответсвующих бензгидрильных катионов позволили получить значения нуклеофильности N и фактора чувствительности, зависящего от природы нуклеофила, (sN) борогидридов в соответствии с Уравнением (1):
log(k2) = sN (N + E) (1)
В ходе данной работы было показано, что HB(2,4,6-F3C6H2)3-Et4N+ и HB(2,6-F2C6H3)3-Et4N+ имеют близкие значения нуклеофильности (N/sN = 14.04/0.61 и N/sN = 13.97/0.66, соответственно), а HB(C6F5)3-Et4N+ оказался гораздо менее реакционно-способным (N/sN = 10.01/0.71). Кинетические исследования реакций HB(2,6-F2C6H3)3- с различными противоионами: Et4N+, n-Bu4N+, 2,2,6,6-тетраметилпиперидиниум (TMPH+), t-Bu3PH+ и (i-Pr)2EtNH+ – и бензгидрильной соли бис(1-метилиндолин-5-ил)метилиум тетрафторбората показали практически одинаковые значения k2. Исходя из этих результатов, можно сделать вывод о том, что водородная связь между протонным и гидридным центрами в борогидридах не влияет на нуклеофильность гидридных центров.
Отношение значений k2 реакций HB(2,6-F2C6H3)3-Et4N+ и HB(2,6-F2C6H3)3-TMPH+ с 2,6-дифенил-4-(4-метоксибензилиден)циклогекса-2,5-диеноном меньше двух (5.5 и 10.8 M ?1с?1 соответственно), что пренебрежительно мало, и мы можем заключить, что активации электрофила путем образования водородной связи с протонами катиона TMPH+ не происходит. Мы также исследовали кинетику реакций борогидридов с другими электрофилами. Значения k2, которые были рассчитаны по уравнению (1) с помощью измеренных N and sN параметров, совпадают с экспериментально полученными k2 для реакций борогидридов с хинон метидами и иминиевыми солями. Однако экспериментальные значения k2 для реакций борогидридов с некоторыми акцепторами Михаэля, такими как бензилиден кислотами Мельдрума и бензилиден-малононитрилами оказались практически в 100 раз медленнее, чем рассчитанные по уравнению (1).
В качестве выводов данной работы можно сказать, что, во-первых, мы разработали альтернативный метод синтеза РПЛ-H2 солей без использования H2, который позволил нам получить различные аммониевые и фосфониевые борогидриды с высокими выходами в мягких условиях. Полученные данные о реакционной способности (N и sN) свидетельствуют о том, что использование менее электрофильных боранов приводит к образованию более нуклеофильных борогидридов HB(Ar)3-. Из полученных N параметров и уравнения (1) мы можем предсказать круг потенциальных реагентов (иминиевые соли, акцепторы Михаэля) с аккуратностью до 2 порядков реакционной способности. Мы также показали, что наличие водородной связи в борогидридах не влияет на их способность быть донорами гидридов, и что карбонильные группы хинон метидов в реакциях с боргидридами не активируются водородной связью с протонами противокатиона.
Список литературы
[1] D. W. Stephan, G. Erker, Angew. Chem. Int. Ed. 2010, 49, 46–76.
[2] J. Paradies, Angew. Chem. Int. Ed. 2014, 53, 3552–3557.
[3] J. Nicasio, S. Steinberg, B. Ines, M. Alcarazo, Chem. Eur. J. 2013, 19, 11016–11020.
[4] L. Greb, C.-G. Daniliuc, K. Bergander, J. Paradies, Angew. Chem. Int. Ed. 2013, 52, 5876–5879.
[5] H. Mayr, M. Patz, Angew. Chem. Int. Ed. 1994, 33, 938–957.
Список публикаций
Guillaume Berionni, Varvara Morozova, Maximilian Heininger, Peter Mayer, Paul Knochel, and Herbert Mayr Electrophilic Aromatic Substitutions of Aryltrifluoroborates with Retention of the BF3— Group: Quantification of the Activating and Directing Effects of the Trifluoroborate Group // Journal of the American Chemical Society, 2013,135,6317-6324Синтез пентафторэтильных комплексов меди (I) и их использование в реакции пентафторэтилирования хлорангидридов карбоновых кислот
Институт химических исследований Каталонии; Institut Catala d'Investigacio Quimica (ICIQ), Таррагона, Испания
Научный руководитель: профессор , к. х.н.
Ранее в нашей лаборатории был открыт новый способ синтеза реактива пентафторэтил меди “CuC2F5”.
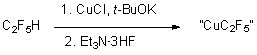
В настоящей работе из данного реактива был получен и структурно охарактеризован ряд пентафторэтильных комплексов меди (I).
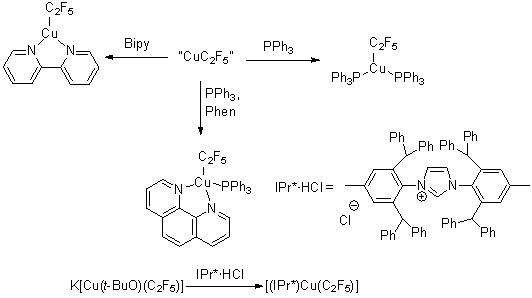
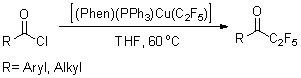
Далее полученный комплекс [(Phen)(PPh3)Cu(C2F5)] был введен в реакцию пентафторэтилирования с серией ароматических и алифатических хлорангидридов карбоновых кислот.
Селективность, активность и стабильность биметаллических катализаторов Pd-Mе в гидрировании замещенных алкинов
ИОХ РАН имени
Научный руководитель: д. х.н.
Селективное гидрирование алкинов до алкенов является одной из базовых реакций в современной органической химии. Получаемые в ходе реакции алкены широко используются в производстве биологически активных веществ: пищевых добавок, витаминов, лекарственных средств [1,2]. С другой стороны, селективное гидрирование тройной углерод-углеродной связи до двойной является важнейшей стадией очистки от ацетиленовых примесей промышленного сырья, используемого в процессах полимеризации [3].
С технологической точки зрения наиболее эффективными являются гетерогенные моно - и биметаллические Pd-содержащие системы [4,5]. Использование биметаллических ацетатных комплексов Pd в качестве предшественника позволило получить высокоселективные катализаторы очистки этилена от ацетиленовых примесей [6]. Высокая селективность полученных образцов обусловлена гомогенностью Pd-M сплавных наночастиц, образующихся в ходе их восстановительной активации.
Рис.1 Схема реакции гидрирования ДФА
Целью данной работы является изучение каталитических характеристик биметаллических нанесенных Pd-M систем, приготовленных на основе гетеробиметаллических ацетатных комплексов, в жидкофазном гидрировании дифенилацетилена (ДФА).
В качестве второго металла использовали Co, Zn, Ni и Ag. Установлено, что введение второго металла несколько снижает активность катализатора (Pd > Pd-Co > Pd-Zn > Pd-Ni > Pd-Ag). Однако существенным преимуществом биметаллических катализаторов является их более высокая селективность (88-90%) в образовании цис-стильбена практически во всем интервале конверсий ДФА (~20-85%). Более явно различия в селективности моно - и биметаллических катализаторов проявляются при высоких конверсиях ДФА (85-90%). Следует отметить, что введение второго металла увеличивает и стабильность работы катализатора.
Ссылки
, , Российские нанотехнологии, том 4, 9-10 (2009), 94 P. C. L’Argentiere, M. E. Quiroga, D. A. Liprandi, E. A. Cagnola, J. A. Diaz-Aunon, M. C.Roman-Martinez, C. Salinas-Martinez de Lecea, Catal. Lett. 87 (2003) 97–101 Cheung T.-T. P., Selective hydrogenation catalyst and processes therefore and therewith // U. S. Patent 7 009 085, Publ. 2006. P. Albers, J. Pietsch, S. F. Parker, Journal of Molecular Catalysis A: Chemical 173 (2001) 275–286 A. Tungler, G. Fogassy, Journal of Molecular Catalysis A: Chemical 173 (2001) 231–247 I. S.Mashkovskii, O. P.Tkachenko, G. N.Baeva, A. Yu. Stakheev. Kinetics and Catalysis. 50, 768 (2009) [Кинетика и катализ, 50, 798 (2009)]Список публикаций:
1. I. S. Mashkovsky, A. V. Sergeeva, A. S. Kashin, D. S. Krivoruchenko, N. Yu. Kozitsyna, M. N. Vargaftik, A. Yu. Stakheev, I. I. Moiseev, Bimetallic Pd-Zn/Al2O3 catalyst in selective diphenylacetylene semi-hydrogenation // Submitted to Mendeleev Communications.
Тезисы докладов на конференциях
1.? I. S. Mashkovsky, A. V. Sergeeva, A. S. Kashin, A. Yu. Stakheev, Stereoselective semihydrogenation of diphenylacetylene over supported heterogeneous Pd catalysts: effect of the support and the second element, UK – Russia Frontiers of Science Symposium
2.? I. S. Mashkovsky, A. V. Sergeeva, A. S. Kashin, D. S. Krivoruchenko, N. Yu. Kozitsyna, M. N. Vargaftik, A. Yu. Stakheev, Effect of Alloying and Preparation Methods on the Performance of Pd-supported Bimetallic Catalysts in Stereoselective Semihydrogenation of Diphenylacetylene 6th International Conference on Green and Sustainable Chemistry (GSC-6), August 4-7 2013, Nottingham, UK
Ареновые комплексы рутения с аминоиминофосфоранатными лигандами: синтез, строение и некоторые каталитические свойства
ИНЭОС РАН имени
Научный руководитель: д. х.н. и
Химия гетероатомных аминоиминофосфоранатных комплексов (NPN) металлов, особенно поздних переходных металлов, развита слабо. Например, в литературе описаны чуть более десятка примеров NPN-комплексов поздних переходных металлов и лишь один для рутения.1 Это связано с малой изученностью свойств самого NPN-лиганда и отсутствием эффективных методов их синтеза. Недавно в лаборатории МОС ИНЭОС РАН были разработаны удобные методы синтеза диаминофосфониевых солей, являющихся предшественниками NPN-лигандов2 и было показано, что в комплексах NPN-лиганды имеют цвиттер-ионную структуру.3 Целью настоящего исследования являлась разработка методов синтеза серии новых комплексов рутения с NPN-лигандами, изучение их строения и каталитических свойств. Для этого были синтезированы три диаминофосфониевые соли с различными заместителями при атомах фосфора и азота R2P(NHR’)2+Br - (R = Ph, Et; R’ = p-Tol, Me) и предложены удобные методы синтеза 18e и 16e комплексов рутения с этими лигандами [(Arene)RuCl{R2P(NR’)2}] и [(Arene)Ru{R2P(NR’)2}]+X-. Комплексы обоих типов были изучены методами ЯМР и УФ при различных температурах, а их строение установлено методом рентгеноструктурного анализа. На основании полученных данных установлено, что NPN-лиганд является сильным 4- или 6-электронным донором и способен эффективно стабилизировать электронно-ненасыщенные (Arene)Ru фрагменты. Установлено, что нейтральные хлоридные 18e комплексы способны диссоциировать в органических растворителях с разрывом связи Ru-Cl и образованием формально 16e катионных соединений. Этот процесс протекает тем легче, чем выше донорная способность заместителей в NPN-лиганде. Методами ЯМР и УФ были определены константы диссоциации и энергии активации процесса разрыва-образования связи Ru-Cl. Для одного из комплексов получены равновесные термодинамические параметры (?H, ?S), что позволило сделать предположение о механизме этого процесса. Кроме того, изучено взаимодействие NPN-комплекса с различными n-донорными лигандами (MeCN, Py, CO), подтверждающие высокие ?- и ?-донорные способности NPN-лиганда.

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 |
