
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Альтернативный сплайсинг вызваный MDX3
Поскольку меньшие транскрипты избирательно амплифицированны ПЦР, интенсивность связок на гелях не отражает относительное изобилие транскриптов в клетке (см. Рис. 6B).. Это на первый план выдвигало факт, что продукт во всю длину всегда одинаково амплифицируются когда внутренние инициаторы в пределах 1-2 КБ друг от друга (например. F19/R32 в Рис. 6B), использовались в реакции ПЦР. С внутренними инициаторами, все более и более отдаленными от друг друга (например. F9/R43in или F9/R50in), только альтернативные транскрипты были усилены. Это не происходило из-за неполного ретротранскрипции, так как транскрипт во всю длину всегда одинаково усиливался, когда мы использовали пары инициатора F22/R32 (данные, не показанны). Поэтому, этот тип анализа не обеспечивает данные, которые точно отражают уровни альтернативных транскриптов относительно транскрипта во всю длину.
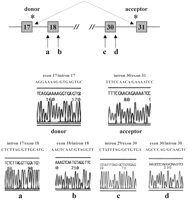
Неожиданное обнаружение многократных разновидностей сайтов сплайсинга, вызванных единственной изменением основания в соединении, принудило нас исследовать другие сайты сплайсинга, специфично донорские и акцепторные места для экзонов, которые заключали пропущенные экзоны для мутаций. Мы не ожидали это по двум главным причинам. Во-первых, не было никакого соответствия между последовательностями других сайтов сплайсинга и интрон 22/экзон 23 соединением. Таким образом не было бы никакого гомологичного соединения между MDX3 и ни одним из тех сайтов сплайсинга, которые могли бы привести к таким изменениям. Во-вторых, MDX4 почти идентичен MDX3 и все же не вызывал дополнительного соединения. Таким образом это было бы необычно, если бы MDX3 был способен к стимулированию мутаций на многократных, негомологичных сайтах сплайсинга вдоль гена дистрофина, тогда как у MDX4, отличающегося только единственным основанием от MDX3, не было никакой активности вообще в стимулировании мутаций. Однако, основанный на замене структуре соединения, идентифицированного в MDX3-инфицированных-вирусом клетках (Таблица 1), мы исследовали другие сайты сплайсинга на мутации. Мы PCR-амплифицировали донорский сайт сплайсинга 5 ′ экзонов и акцепторной сайт сплайсинга для 3 ′ экзонов для каждого нового соединения отдаленных экзонов друг с другом. Кроме того, мы упорядочивали и донорские и акцепторные сайты сплайсинга для первого 'пропущенного' экзона или от 5 ′ или от 3 ′ (см. Рис. 7 для примера). Вся амплификация была пройдена в два раунда перед отщеплением рисунке 2C. Рисунок 7 показывает данные о последовательности для случая, в котором было добавочное соединение между экзоном 17 и экзоном 31 (продукт ‘d’ от Рис. 6 и Таблицы 1). Не было никакого признака изменения оснований в последовательности сайтов сплайсинга или смежных последовательностях ни для одного места, даже после двух раундов амплификации ПЦР. Идентичный анализ был сделан для продуктов ‘b’, ‘c’, ‘e’ и ‘f’ (Рис. 6 и Таблица 1), ни один из которых не приводил доказательства мутаций сайтов сплайсинга. Поэтому, единственная мутация, идентифицированная в гене дистрофина среди этих 25сайтов сплайсинга, была единственной, в интроне 22/экзон 23 соединении. Эти данные предполагают, что мутации в единственных сайтах сплайсинга могут иметь более широкие эффекты чем пропуск единственного экзона в соединении сложного гена как дистрофин.

Большая версия изображения:
In this page
In a new window
Download as PowerPoint Slide
Рисунок 7.
Дискуссия
Мы расширили терапевтический потенциал химеропласт - опосредованной замены основания от простой коррекции точечных мутаций до восстановления рамки считывания. Этот подход может быть применим для мутаций с изменением рамки считывания (включая большие делеции) У людей этот метод потенциально должен способствовать преходу фенотипа МДД в более мягкий фенотип МДБ.. Схожий результат является целью антисмыслового-опосредованного пропуска экзона, что применено к мутациям гена дистрофина у людей и мышей (8,10–12). Пока антисмысловой-опосредованный пропуск экзона дает надежду, использование химеропласта для пропуска экзона в гене дистрофина имеет некоторые приемущества. Во-первых, коррекция с использованием химеропласта происходит на геномном уровне, что освобождает от необходимости продолжительной доставки олигонуклеатидов или экспрессии антисмысловой конструкции для поддержания экспрессии дистрофина. Во-вторых, генная коррекция может быть применима не только к зрелым имофибриллам, (21) но и их предшественникам (20), которые участвуют в репарации мышц.. Таким образом терапевтическая выгода была бы устойчива не только с точки зрения исправления на геномном уровне, но также и с точки зрения оборота в ткани, поскольку клетки предшественника мышцы соединяются с существующими мышечными волокнами или друг с другом, чтобы сформировать новые мышечные волокна. В настоящее время эффективность основного преобразования - по крайней мере, порядок величины ниже, чем было бы необходимо для терапевтического применения, и это остается одним из главных препятствий для этого типа технологии генотерапии.
Наши результаты демонстрируют, что нацеленный химеропласт(MDX3) способен к стимулированию преобразования G-T в интрон 22/экзон 23 соединении гена дистрофина (Рис. 1B), и, таким образом, разрушить соединение экзона 22 к экзону 23. Однако, это геномное преобразование имело более широкие воздействия на соединении премРНК дистрофина и заканчивалось экспрессией многократных форм белка, некоторых с большими внутренними делециями. Анализ МРНК подтвердил наличие альтернативно соединенных транскриптов с числом исключенных экзонов в пределах от 11 - 27 (Рис. 6). Вестерн-блот-анализ используя антитела для С - терминальной области дистрофина показывал наличие по крайней мере трех различных белковых продуктов (Рис. 3A). Иммунное окрашивание используя антитела, определенные для различных частей дистрофина, подтвердило, что у произведенных белков были внутренние делеции.(Рис. 3C).
Представленные данные, привели к неожиданному результату- мутация в единственном основании в одном сайте сплайсинга могла изменить сплайсинг более глобально в пределах гена дистрофина. Последовательность химеропласта, выбранная для этого эксперимента, не соответствовала никаким другим геномным последовательностям, таким образом гарантируя специфичность олигонуклеатидов. Изменение даже единственного основания в химерном олигонуклеатиде резко уменьшает свою склонность вызвать преобразования (23). Таким образом не было никакой причины ожидать, что химеропласт, предназначенный к соединению интрона 22/экзона 23, будет предназначаться для негомологичногосайта сплайсинга. Было показано, что химеропласт, предназначенный для определенных геномных последовательностей способен к стимулированию одно-основных преобразований в генах, но не имеет никакой мутационной склонности даже в гене с очень гомологичной последовательностью (24). Таким образом, хотя мы не можем полностью исключить возможность, что у MDX3 могла быть мутационная активность в другом месте генома, очень маловероятно, что это вызывает преобразования специфично в сайтах сплайсинга в гене дистрофина. Однако, чтобы поддержать эти теоретические аргументы, мы непосредственно упорядочили все поврежденные сайты сплайсинга и нашли признак преобразования только в предназначенной основании (Рис. 2C и 7). Кроме того, как отмечено ранее, любая гипотеза, что замена, вызванная MDX3, происходит из-за мутаций большего количества сайтов чем только единственное основание в интрон 22/экзон 23 соединении, должна была бы принять во внимание факт, что у MDX4, идентичного MDX3 кроме единственного изменения пары оснований, не было никакой склонности вызвать дополнительное изменение гена.

Хотя MDX3 мог теоретически иметь эффект на РНК, обрабатывающую непосредственно (например, активностью антисенсорного типа), возможность, что альтернативные продукты явились результатом постоянно действующего на обработку РНК химеропласта, исключена нашими долгосрочными результатами. Клетки распространяются с прериодом удвоения приблизительно 18 часов. Мы видим ту же самую структуру транскрипта и белковой экспрессии спустя 3 месяца после трансфекции, как мы делаем в течение 1 недели после трансфекции. В 3-месячном отрезке времени количество химеропласта на клетку уменьшилось бы фактором 2120 со времени трансфекции, основанной исключительно на разбавлении, которое будет встречаться во время клеточного деления. Это соответствует 1Ч1036-кратному сокращению концентрации химеропласта и даже не принимает во внимание деградацию химеропласта в клетках, который, вероятно, будет иметь, по крайней мере, такойже эффект на сокращение химеропласта на клетку, как клеточное деление. Таким образом количество химеропласта в клетке в 3-месячном отрезке времени было бы настолько бесконечно мало, чтобы быть действительно незначительным. Мы поэтому приходим к заключению, что ни один из результатов не может быть объяснен введенным химеропластом.
Неясно, почему изменение единственного сайта сплайсинга на геномном уровне заканчивается экспрессией многократных продуктов, каждый из которых пропускает много экзонов и вверх по течению и вниз по течению предназначенного сайта сплайсинга. Этот феномен однако наблюдался в клинических случаях пациентов с МДД, в которых точечные мутации в местах сайтов сплайсинга приводят к пропуску многократных экзонов в гене дистрофина. (29,30). Изменение специфической интрон/экзонной последовательности, содержащей регулирующие элементы для построения факторов сплайсинга может разрывать вторичную структуру необходимую для координации сплайсинга продуктов множественных экзонов.(31).Конечная м РНК может продемонстрировать пропуск множественных экзонов расположенных выше и ниже точечной мутации Действительно пропуск смежных экзонов наблюдается когда процесс сплайсинга разрушен поставкой антисмысловых нуклеатидов. (9).

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 |
