
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral

Восстановление экспрессии дистрофина в клетках mdx мышей при помощи химеропласт - опосредованного пропуска экзона.
Carmen Bertoni1, Catherine Lau1 and Thomas A. Rando1,2,*+ Author Affiliations
1Department of Neurology and Neurological Sciences, Stanford University School of Medicine, Stanford, CA, USA and 2GRECC, VA Palo Alto Health Care System, Palo Alto, CA, USA- Received December 15, 2002. Accepted March 18, 2002
В большинстве случаев мутации приводящие к миодистрофии Дюшена представлены большими делециями, что приводит к сдвигу рамки считывания. В результате в мышцах таких индивидов отсутствует или значительно снижено содержание дистрофина Любое лечебное воздействие, которое может восстановить рамку считывания, потенциально способно уменьшить серьезность болезни, позволяя производить частично функциональный дистрофин, как встречается при мышечной дистрофии Беккера (МДБ). Один подход к восстановлению рамки считывания должен был бы изменить соединение пре мРНК, чтобы произвести изменения в структуре. Мы проверили способность РНК/ДНК олигонуклеатидов (химеропластов) изменить ключевые основы в определенных последовательностях в гене дистрофина, чтобы вызвать пропуск экзона. Используя клетки mdx мыши как модель болезни, мы показываем, что химеропласт-опосредованное преобразование в соединении интрон22/экзон 23 вызывает альтернативное соединение и производство транскриптов. Действительно, множественные формы дистрофина наблюдались вестерн-блот-анализом, и функциональность продуктов была продемонстрирована восстановлением экспрессии и локализацией дистрофин-связанного белка, б-дистрогликана, в дифференцированных клетках. Эти данные демонстрируют, что химеропласты могут вызвать пропуск экзона, изменяя последовательности места соединения на геномном уровне. Также, у химеропласт-опосредованного пропуска экзона есть потенциал, который будет использоваться, чтобы преобразовать тяжелый фенотип МДД в намного более умеренный фенотип МДБ.
Предисловие
Мышечная дистрофия Дюшена (МДД) и Беккера (МДБ) являются болезнями мышц с Х сцепленным рецессивным типом наследования, вызванными дефектами в гене на 2.5 МБ, который кодирует белок дистрофин. (1). Больше чем у 80 % пациентов с МДД или МДБ есть большие делеции в гене дистрофина, дупликации, маленькие делеции, вставки и точечные мутации составляют остальные случаи (2). В общем тяжелый фенотип МДД вызван мутациями, такими как делеции сдвига рамки считывания и точечные мутации, вызывающие дефицит дистрофина. Более умеренный фенотип МДБ обычно коррелирует с делециями, которые сохраняют рамку считывания и производит внутренне измененый, но частично функциональный, дистрофин(3). Таким образом у любого лечебного воздействия, которое могло преобразовать мутацию со сдвигом рамки считывания в мутацию без сдвига рамки считывания, есть потенциал, чтобы преобразовать тяжелый фенотип МДД в более умеренный фенотип МДБ.
Наиболее широко изученные стратегии по восстановлению экспрессии дистрофина у дистрофин дефицитных мышцей используют вирусную поставку функциональной комплементарнойой ДНК, кодирующей дистрофин (4–7). Этот подход был эффективен на моделях животных и дает надежду для применения у людей. Кроме того этот подход применим к лечению любого типа мутации МДД, не только делецию сдвига рамки считывания. Однако, вирусные системы доставки сталкиваются с трудностями, такими как врожденный иммунный ответ против векторов, проблемы, связанные с вирусной интеграцией в геном, и адекватным регулированием экспрессии поставленного гена. Несколько групп исследователей изучают возможность использования антисмысловых олигонуклеотидов, чтобы изменить обработку транскрипта дистрофина, и вызвать пропуск экзона и мутации со сдвигом рамки считывания перевести в мутации без сдвига рамки считывания (8–12). Эта технология избегает некоторых из препятствий вирусной опосредованной генотерапии. Однако, этот подход потребовал бы повторенной поставки олигонуклеотидов, чтобы поддержать экспрессию дистрофина, или возникнет потребность в длительной экспрессии антисмыслового транскрипта, как могло бы быть достигнуто с вирусной системой доставки (8). Привлекательная альтернатива антисмысловому пропуску экзона восстановление трансляционной рамки считывания, которая была бы генной стратегией модификации. Текущие генные стратегии репарации, которые могли быть применены к этой проблеме, включают технологии, вовлекающие химерные олигонуклеатиды, одноцепочечные ДНК олигонуклеатиды, и гомологичные короткие фрагменты (13,14).
Генная репарация, вовлекающая химерные РНК/ДНК олигонуклеатиды (химеропласты), основана на активности этих векторов вызвать преобразования пары оснований на геномном уровне (15). Их механизм действия вовлекает использование врожденной репарации в клетке, направляя замену единственного основания в определенной последовательности ДНК (16,17). Химеропласт-опосредованная генная репарация была успешно применена к различным типам клеток, и in vitro и в естественных условиях (18–25). Хотя прежде всего изучено применение для того, чтобы восстановить точечные мутации, у химеропласт-опосредованных последовательностей также есть потенциал, чтобы изменить соединение и таким образом восстановить рамку считывания в гене с мутацией со сдвигом рамки считывании
Хотя прежде всего изучено применение для того, чтобы восстановить точечные мутации, у химеропласт-опосредованных преобразований последовательностей сайтов сплайсинга также есть потенциал, чтобы изменить сплайсинг и таким образом восстановить трансляционную рамку считывания в гене с мутацией со сдвигом рамки считывания.
В этом отчете мы исследовали возможность использования химеропластов, чтобы изменить сплайсинг гена дистрофина. У образцовой системы, mdx мыши, есть мутация в экзоне 23 гена дистрофина, которая ведет к отсутствию белка дистрофина. Мы ранее показали, что химеропласты в состоянии исправить эту точечную мутацию и in vitro и в естественных условиях (20,21). Мы проверили, могла ли мутация единственного основания в интроне 22/экзон 23 соединении изменить сплайсинг транскрипта дистрофина с пропуском экзона 23. Предсказанный продукт, в котором экзон 22 непосредственно соединен к экзону 24, произвел бы белок дистрофин почти во всю длину. Хотя у mdx мышей нет делеции сдвига рамки считывания, они обеспечивают возможность заняться исследованиями того, как устойчивые, одно-основные перестройки геномной ДНК могут использоваться, чтобы переадресовать соединение, которое было бы применимо к мутациям со сдвигом рамки считывания в гене дистрофина. Мы нашли, что, изменяя интрон 22/экзон 23 соединения действительно выдают транскрипты в структуре и сокращенные формы белка дистрофина. Эти исследования расширяют терапевтическое применение химеропласт-опосредованных замен единственного основания для делеций сдвига рамки считывания.

Результаты

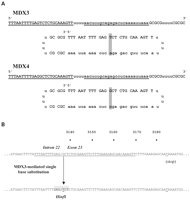
Планирование химеропласта, названного MDX3 (Рис. 1A), было разработано, чтобы вызвать преобразование G-T в экзоне 23 места акцептора интрона 22/экзон 23 соединения в гене дистрофина мыши (Рис. 1B). Изменеие основания, на которое нацелен MDX3, закончилась бы разрушением интрона 22/экзон 23 последовательности и создало бы новый сайт рестрикции в этом положении (Рис. 1B). Предсказанная последовательность транскрипта в результате соединения экзона 22 с экзоном 24 была бы в рамке и приводила к синтезу белка на 71 аминокислоту короче чем дистрофин во всю длину. Как контроль, мы использовали химерный олигонуклеотид, MDX4 (Рис. 1A), разработанный совершенно комплементарным к интрону 22/экзон 23 и таким образом не имеющий никакой активности и не вызывающий изменение геномной последовательности
Большая версия изображения
In this page
In a new window
Download as PowerPoint Slide
Рисунок 1
Признаки химеропласт-опосредованной замены одного основания на геномном уровне
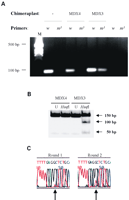
Специфичность MDX3 вызвать преобразование G-T в интроне 22/экзоне 23 была оценена, используя пробу ARMS геномной ДНК. Клетки были поддержаны в культуре в течение по крайней мере 15 дней после трансфекции химеропласта. Используя инициаторы ПЦР, разработанные, чтобы усилить последовательность, имеющую трансверсию G-T, мы наблюдали определенный продукт амплификации только в клетках, инфицированных вирусом с целевым химеропластом (Рис. 2A). Клетки, инфицированные вирусом с контрольным химеропластом или неинфицированными вирусом клетками, были не в состоянии произвести любой продукт амплификации, используя эти инициаторы.
Большая версия изображения
In this page
In a new window
Download as PowerPoint Slide
Рисунок 2..
Преобразование G-T целевым химеропластом было также подтверждено прямым усвоением продуктов амплификации ПЦР(Рис. 2B). Инфицированные вирусом клетки были поддержаны в культуре больше 45 дней, затем геномная ДНК была извлечена. Определенные инициаторы ПЦР использовались, чтобы произвести продукт амплификации 150 bp, которые содержали интрон 22/экзон 23 соединения, которые были подтверждены прямым отщеплением. После очистки продукты ПЦР были подвергнуты перевариванию, используя Hinf I эндонуклеазы. Продукты переваривания в ожидаемом размере ∼50 и 100 bp наблюдались от клеток, инфицированных вирусом с MDX3 (Рис. 2B), демонстрируя, что нацеленые химеропласты вызвали единственную замену пары оснований.

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 |
