
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Если процессы термического восстановления доминируют, тогда Тс определяется из выражения:
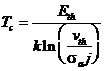 , (1.109)
, (1.109)
где sa — основное сечение для процессов, приводящих к аморфизации вблизи Тс (чаще всего это будет сечение прямой аморфизации). Таким образом, если термовосстановление доминирует, Тс зависит от скорости повреждения sa j, которое для sa j > 1 сдвигает Тс в область более высокой температуры, чем температура, вычисляемая из выражения Eth /[kln(uth)]. Более того, в большинстве экспериментов по ионной аморфизации кристаллов условия выбираются такими, что скорость генерации нарушений sa j была приблизительно постоянной для различных ионов. Критическая температура Тс при таких условиях будет независимой от массы ионов, если доминируют процессы термического восстановления.
Такой тип характеристики наблюдается в случае кристаллов Cd2Ti2O7 [129] (рис. 1.19), а также ZrSiO4 [129]. Предполагая sa постоянным, может быть определено влияние плотности ионного тока j по сдвигу в Tc.Если же доминируют радиационно-ассистируемые процессы восстановления решетки, тогда Tc определяется из выражения:
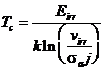 . (1.110)
. (1.110)
Учитывая, что virr = sr j, получим:
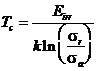 . (1.111)
. (1.111)
При этих условиях, Тс не зависит от j и обратно пропорционально sr /sa. Для оценок можно считать, что это отношение представляет собой отношение числа
свободно мигрирующих дефектов к числу атомов в аморфном кластере, создаваемом одним ионом. Если sr £ sa, что соответствует случаю тяжелых бомбардирующих ионов, Тс не будет зависеть от процессов радиационно-ассистируемого восстановления.
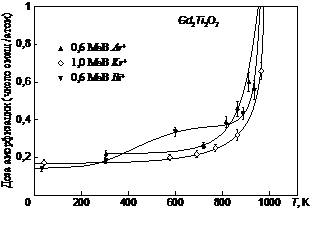 |
Рис.1.19. Температурная зависимость дозы аморфизации Cd2Ti2O7 различными ионами [129].
Температура рекристаллизации аморфных слоев часто зависит от массы бомбардирующих ионов. Так, в случае аморфизации a-кварца ионами щелочных металлов Cs+, Na+ и Li+, температура рекристаллизации на воздухе уменьшается с уменьшением массы ионов (Cs+: 875°С; Na+: 800°С; Li+: 650–700°С) [144]. a-кварц, имплантированный собственными (Si+ или O+) ионами, не рекристаллизуется эпитаксиально при отжигах до температур ~1050°С [145]. Предполагается [144], что активными дефектами, ответственными за твердофазный эпитаксиальный рост, являются Si–O‑связи, ослабленные наличием сетки модифицирующих щелочных металлов.
Низкотемпературная аморфизация в материалах, где диэлектрический отжиг подавлен, может быть описана в рамках модели плотных каскадов столкновений, генерируемых налетающими ионами в мишени. К таким материалам следует отнести a-кварц, основным механизмом аморфизации которого при этих температурах является образование и рост локальных разупорядоченных областей [146]. При повышенных температурах одновременно имеет место аморфизация и динамический отжиг. С ростом температуры последний процесс становится доминирующим. Модель диффузии вакансий из ядра каскадной области, предложенная Морехедом и Краудером [98], является адекватной для описания этих условий [147]. Критический флюенс для аморфизации Dc ионами Ne+ с Е = 50 кэВ не зависит от температуры до Т = 550 К при ионном облучении a-кварца, однако с ростом температуры он резко увеличивается (рис. 1.20). Критическая температура для проявления аморфизации при ионном облучении составляет около 940 К [147]. Энергия активации для диффузии дефектов в каскадной обласили на интерфейсе аморфная фаза/кристалл составляет 0,28 ± 0,02 эВ.
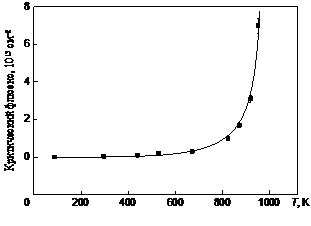 |
Рис. 1.20. Критический флюенс Dc как функция температуры кристаллов a-кварца: сплошная линия — модель Морехеда–Краудера [98] с Еа = 0,28 эВ, Тс = 986 К; квадраты — эксперимент [147]
Флюенс для аморфизации зависит от типа ионов. Например, ионы циркония вызывают аморфизацию Al2O3 (корундовая керамика) при флюенсе в два раза меньшем, чем при облучении ионами ниобия с той же энергией [148]. Облучение пленок корундовой керамики ионами Zr+, Cu+ и Ti+ [149] c дозами от 1017 до 2´1017 см–2 и энергией 170 кэВ выявило аморфизацию мишени в первом случае, трансформацию a-корунда в g‑корунд во втором и в d-Al2O3 в третьем случае. Постимплантационные отжиги при Т = 600–1000°С вызывают возврат решетки к равновесному состоянию путем кристаллизации a-Al2O3 и преципитации оксидов имплантированных атомов (ZrO2, CuO и CuAl2O4, а также TiO2) [149].
При облучении сапфира (монокристаллический Al2O3) ионами Xe3+, Xe2+, Kr2+, Ar2+, O+ при ускоряющем напряжении 300 кэВ и температурах от 90 до 230 К, обнаружена [150] аморфизация ионами ксенона и криптона с Е = 600–900 кэВ и отсутствие аморфизации при облучении легкими ионами аргона и кислорода. Флюенс для аморфизации составлял 3 и 4 смещ./атом соответственно для ионов Xe и Kr при температурах от 90 до 160 К. Затем Dс возрастает до 8 смещ./атом, далее увеличиваясь экспоненциально при Т > 200 К. Пороговая температура для аморфизации зависит от плотности ионного тока и массы ионов, хотя для Т < 200 К зависимость от j не наблюдалась. Сапфир оставался в кристаллическом состоянии при Т > 250–300 К. В случае облучения поликристаллических пленок Al2O3, критическая доза Dc при облучении ионами Xe с Е = 900 кэВ при 90 К возрастает до 3,8 смещ./атом, т. е. наличие границ зерен — стоков для точечных дефектов обуславливает увеличение Dc на 40% в указанных условиях [150].
В целом при облучении сапфира тяжелыми ионами методом ПЭМ наблюдалось 4 стадии структурных превращений при Т < 300 К:
1) накопление сильно нарушенных областей вызывает аморфизацию при Т от 90 до 160 К;
2) отжиг близких пар Френкеля внутри каскадов происходит при Т от 160 до 200 К;
3) отжиг точечных дефектов при Т > 200 K приводит к экспоненциальной зависимости Dc(T) и зависимости Тс от j;
4) сохранение кристалличности сапфира при Т > 250–300 K [150].
Кристаллы сапфира с с-осью, параллельной направлению пучка ионов Sn+ c Е = 180 кэВ, аморфизуются при комнатной температуре в интервале флюенсов 5´1015–1´1016 см–2 [151]. Наблюдалась [151] кристаллизация в окисляющей среде при Т < 960° C в следующей фазовой последовательности: аморфный Al2O3 ® g-Al2O3 > a-Al2O3. Ионно-индуцированная аморфизация кристаллических фаз в системе Al2O3–SiO2 (Al2O3; SiO2 (кварц); Al2SiO5 (кианит, андалузит и силлиманит); 3Al2O3×2SiO2 (муллит)) исследовалась при облучении их высокоэнергетичными ионами Xe+ с Е = 1,5 МэВ в интервале температур 15–1023 К [152]. Критические температуры Тс, выше которых не происходила полная аморфизация при данных экспериментальных условиях, были равны: 1446 К для кварца, 1337 К для силлиманита, 1290 К для кианита, 1031 К для андалузита, 616 К для муллита и 123 К для a-Al2O3. Тс‑значения вычислялись на основе модели Вебера и др. [105]. Результаты интерпретировались [152] с учетом схожести процессов радиационно-индуцированной аморфизации и стеклообразования.
В зависимости от типа ионов и их энергии, аморфизация кристаллов SiC (как для 6H–SiC, так и для 3C–SiC) в диапазоне температур, ниже комнатной, происходит при флюенсах выше 0,25–0,63 смещ./атом и энергии, выделенной на атом, превышающей 19–56 эВ/атом [121]. Аморфные слои SiC рекристаллизуются при температурах выше 900°C, однако исходное совершенство решетки не достигается даже при очень высокотемпературных отжигах [121]. Конкуренция между эпитаксиальным ростом от межфазной границы аморфный слой — кристалл и полицентровой кристаллизацией и ростом кристаллитов определяет сложную дефектную структуру с включениями 3С–SiC внутри 6H–SiC-матрицы.
Критическая температура для аморфизации при ионном облучении зависит от величины энергии, передаваемой атомами отдачи на единице длины пути, и измеряется в пределах 400–650 К [121, 153]. При облучении 6H–SiC-кристаллов экстремально высокой дозой ионов углерода (D = 1´1018 см–2, Е = 60 кэВ) при повышенных температурах наблюдается [154] формирование различных фаз: аморфная, обогащенная углеродом фаза при 300°C. Преципитаты графита получены при 600°C, в то время как малые алмазные зерна формируются при 900°C. Эти зерна идеально эпитаксиально сопрягаются с окружающей SiC-матрицей. Механизм формирования алмазов в SiC пока не ясен. Можно предположить, что тетраэдрически координированная решетка SiC, которая сохраняется при высокотемпературном облучении, стимулирует зарождение этой фазы. Кроме того, локальные неравновесные условия в каскаде и (или) напряжения, действующие на атомы углерода, вкрапленные в междоузлия, от окружающей SiC-решетки могут вносить вклад в зародышеобразование алмаза. Веселовски и др. [155] обнаружили преобразование графитных углеродных “луковичек” в алмаз при облучении ионами Ne+ с Е = 3 МэВ при Т = 700–1100°C. Последние объясняли процесс превращения графита в алмаз сжатием углеродных “луковичек”, вызванным атомными смещениями. Очевидно, что в обоих упомянутых случаях температура играет решающую роль.
Цирконолиты являются интересными материалами в качестве основной фазы для иммобилизации избытка оружейного плутония и вредных отходов переработки ядерного горючего [156, 157]. Облучение 6 типов цирконолитов осуществлялось ионами Kr+ c Е = 1 МэВ в диапазоне температур 25–973 К [157]. Все эти цирконолиты (CaZrTi2O7; Ca0,8Ce0,2ZrTi1,8Al0,2O7; Ca0,85Ce0,5Zr0,65Ti2O7; Ca0,5Nd0,5ZrTi1,5Al0,5O7; CaZrNb0,85Fe0,85Ti0,3O7 и CaZrNbFeO7) аморфизуются практически при одном и том же флюенсе (~2´1015–3,9´1015 см–2) при комнатной температуре. С ростом температуры аморфизационный флюенс для каждой фазы становится значительно различным. Критическая температура аморфизации составляла 550 К для CaZrNbFeO7; 590 К для CaZrNb0,85Fe0,85Ti0,3O7; 640 К для CaZrTi2O7; 900 К для Ca0,8Ce0,2ZrTi1,8Al0,2O7; 1000 К для Ca0,85Ce0,5Zr0,65Ti2O7 и 1020 К для Ca0,5Nd0,5ZrTi1,5Al0,5O7. Тенденция в изменении критической температуры указывает на то, что уменьшение содержания кальция увеличивает чувствительность материала к аморфизации.
Флюенс для аморфизации цирконолитов как функция температуры мишени описывается выражением:
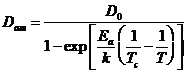 , (1.112)
, (1.112)
где D0 — флюенс аморфизации, экстраполированный к 0 К, Еа изменялось от 0,23 до 0,4 эВ.
Аналогичная (1.112) зависимость Dam(Т) характерна и такому интересному классу изоляторов, как слюды (мусковит, флогопит, биотит, лепидолит), которые облучались ионами Kr+ с Е = 1,5 МэВ [158] в интервале температур 20–1023 К. Все четыре типа слюды оказались весьма чувствительными к ионному облучению. Аморфизационный флюенс составлял всего 1–2´1014 ион/см2 при комнатной и более низкой температурах. Dam увеличивался с ростом температуры кристалла при облучении начиная от Т ³ 400 К. Критическая температура аморфизации Тс для всех четырех слюд была более 1000 К. Лепидолиты {K2(Li, Al)5–6[Si6–7Al1–2O20](OH, F)4} наиболее легко аморфизуемый материал из этих четырех слюд с Тс » 1300 К. Параметры выражения (4.9.23) составляют для мусковита: Еа = 0,08, Тс = 1100 К; для биотита: Еа = 0,09, Тс = 1020 К; для флогопита: Еа = 0,065, Тс = 1000 К; для лепидолита Еа = 0,12, Тс = 1300 К. Аналогичные характеристики для кварца равны: Еа = 0,17, Тс = 1400 К и корунда — Еа = 0,014, Тс = 120 К [158]. Различную чувствительность слюд к аморфизации при ионном облучении Ванг и др. [158, 159] объясняют в рамках модели закалки каскадов (т. е. расплавление с быстрым остыванием). Этот же подход использовался и для случая аморфизации моноалюмината кальция CaAl2O4 [160]. Хотя средняя степень ионности связей в CaAl2O4 составляет 0,68, эта керамика аморфизуется при относительно низком флюенсе ионов Xe+ с Е = 1,5 МэВ при комнатной температуре: 1,2´1014 Xe+/см2. Параметры выражения (4.9.23), описывающие зависимость аморфизационного флюенса от Т, составляют: Тс = 733 К; для биотита: Еа = 0,062 эВ. В то же время шпинель MgAl2O4 весьма устойчивая кристаллическая фаза в отношении аморфизации. Она может аморфизоваться только при флюенсе 25 смещ./атом при Т = 100 К ионами Xe2+ с Е = 400 кэВ [161]. В другом исследовании [162] шпинель не аморфизовалась при флюенсе 15 смещ./атом при Т = 20 К при бомбардировке ионами Kr+ с Е = 1,5 МэВ. Степень ионности межатомных связей в шпинели составляет 0,66, т. е. практически соответствует случаю соединения CaAl2O4. В то же время андалузит, Al2SiO5 с близкой степенью ионности связи (0,59) менее стоек в отношении облучения [160]. Следовательно, критерий аморфизации Нагуиба и Келли [110, 112], предполагающий аморфизуемость материала при условии, когда ионность £0,47, здесь не выполняется. Различие в Тс для этих кристаллов связывается [160] с влиянием тетраэдрических комплексов ([SiO4], [AlO4] и [MgO4]) на стеклообразование, а также различной координацией ближайших соседей алюминия (4 — в CaAl2O4; 5 и 6 — в андалузите и 6 — в шпинели).
Литература к главе 1.
1.
2.
3.
4.
5.
6.
7.
8. E. A.Kotomin, A. I.Popov/ Radiation-induced point defect in simple oxides//Nuclear Instruments and Methods in Phys. Res.,1998, B141, P.1-15.
9.S. B. Ubizskii, A. O.Matkovskii, N. A.mironova-Ulmane/ Radiation displacement defect formation in some complex oxide crystals// Nuclear Instruments and Methods in Phys. Res., 2000, B166-167, P.40-46.
10.R. E.Williford, R. Devanathan, W. J. Weber/ Computer simulation of displacement energies for several ceramic materials// Nuclear Instruments and Methods in Phys. Res.,1998, B141, P.94-98.
11. B. Pa
11.R. Devanathan, W. J.Weber, T. Diaz de la Rubia /Computer simulation of a 10 keV displacement cascade in SiC// Nuclear Instruments and Methods in Phys. Res., 1998, B141, P.118-122.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46. Komarov F. F., Novikov A. P., Burenkov A. F. Ion Implantation, Universities Publ. House, Minsk, 1994, 415 pp.
47.. Lindhard J., M. Scharff, H. E.Schiott. Mat.-Fys. Medd. Dan. Vid. Selsk., 1963, v. 33, No 14.
Bariloche, Argentina, N. R.Arista, 1999, 351 pp.
48. Ziegler J. F., Biersack J. P., SRIM: The Stopping and Range of Ions in Matter, Version 96.07 IBM-Research, Yorktown, 1996.
49. Biersack J. Nucl. Instr. Meth., 1999, v. B153, p. 398–409.
50. Christel L. A., Gibbson J. F., Milroe S. J. Appl. Phys., 1980, v. 51, p. 6176–6182.
51. Christel L. A., Gibbons J. F., Milroe S. Nucl. Instr. Meth., 1981, v. 182/183, p. 187–198.
52. Gibbons J. F. Nucl. Instr. Meth., 1987, v. B22, p. 83–86.
53. Lizunov Yu. D., Ryazanov A. I. Radiat. Eff., 1982, v. 60, p. 95–100.
54. , Рязанов , 1987, N 5, с. 121–131.
55. Burenkov A. F., Komarov F. F., Temkin M. M., Schlotzhauer G. Radiat. Eff. Let., 1984, v. 86, p. 161–168.
56. , , Темкин , 1989, N 4, с. 15–21.
57. Lindhard J., Nielsen V., Scharff M. Mat.-Fys. Medd. Dan. Vid. Selsk., 1968, v. 36, No. 10.
58. Johnson W. S., Gibbons J. F. Projected Range Statistics in Semiconductors, Stanford, California: Stanford University Book Store, 1966, p. 390.
59. Gibbons J. F., Johnson W. S. Mylroie. Projected Range Statistics, Stroudsburg, Pennsylvania: Dowden and Hutchinson and Ross, Inc., 1975, 421 p.
60. Brice D. K. Ion Implantation Range and Energy Deposition Distribution. New Jork: JFJ/plenum, 1975, v. 2, 596 p.
61. Winterbon K. B. Ion Implantation Range and Energy Deposition Distribution. New York: JFJ/plenum, 1975, v. 1, 341 p.
62. , , Темкин пространственного распределения ионно-имплантированных примесей. Минск, изд-во «Университетское», 1980, 352 c. (translated by Gordon and Breach, New York-London-Paris, 1986, 462 p.)
63., , Темкин распределение энергии, выделенной в каскаде атомных столкновений в твердых телах. Москва, Энергоатомиздат, 1985, 248 c.
64. Hofker W. K., Oesthock D. P., Kolman N. J., de Grefte H. A. Radiat. Eff., 1975, v. 24, p. 223–231.
65. Thompson D. A., Robinson J. E., Walker R. S. Radiat. Eff., 1977, v. 32, p. 169–175.
97. Gibbons J. F. Proc. IEEE, 1972, v. 60, p. 1062–1066.
98. Morehead F. F., Crowder B. Z. Radiat. Eff., 1970, v. 6, p. 27–31.
99. Dennis J. R., Hale E. B. J. Appl. Phys., 1978, v. 49, p. 1119–1122.
100. Webb R. P., Carter G. Radiat. Eff., 1981, v. 59, p. 69–72.
101. Carter G. Radiat. Eff., 1986, v. 100, p. 281–285.
102. Hecking N., Heidemann K. F., te Kaat E. Nucl. Instr. Meth., 1986, v. 15B, p. 760–764.
103. Jackson K. A. J. Mater. Res., 1988, v. 3, p. 1218–1221.
104. Weber W. J., Wang L. M. Nucl. Instr. Meth., 1994, v. 91B, p. 63–66.
105. Weber W. J., Ewing R. S., Wang L. M. J. Mater. Res., 1994, v. 9, p. 688–691.
106. Hobbs L. W. Nucl. Instr. Meth., 1994, v. 91B, p. 30–42.
107. Meldrum A., Zinkle S. J., Boatner L. A., Ewing R. S. Phys. Rev., 1999, v. 59B, p. 3981–3986.
108. Wang S. X., Wang L. M., Ewing R. S., in: Atomistic Mechanisms in Beam Synthesis and Irradiation of Materials (eds.: Barbour J. C., Roorda S., Ila D., Tsujioka M. Mater. Res. Soc. Proc. 504, MRS, Warrendale, PA, 1998, p. 165–169.
109. Matzke H. J. Radiat. Eff., 1982, v. 64, p. 3–8.
110. Naguib H. M., Kelly R., in: recent Advances in Science and Technology of Materials, ed. Bishay A., Plenum, N.-Y., 1974, p. 321–324.
111. Wang L. M., Eby R. K., Janaczek J., Ewing R. C. Nucl. Instr. Meth., 1991, v. 59/60B, p. 395–400.
112. Naguib H. M., Kelly R. Radiat. Eff., 1975, v. 25, p. 1–10.
113. Eby R. K., Ewing R. C., Birtcher R. C. J. Mater. Res., 1992, v. 7, p. 3080–3083.
114. Swanson M. L., Parsons J. R., Hoelke C. W. Radiat. Eff., 1971, v. 9, p. 243–253.
115. Vook F. L.Stein H. J. Radiat. Eff., 1969, v. 2, p. 23–27.
116. Pascucci M. R., Hobbs L. W. J. Physique, 1980, v. 41 (C6), p. 237–240.
117. Pascucci M. R., Hutchison J. L., Hobbs L. W. Radiat. Eff., 1983, v. 74, p. 219–224.
118. Gong W. L., Wang L. M., Ewing R. C., Zhang J. Phys. Rev., 1996, v. 54, p. 3800–3804.
119. Weber W. J., Wang L. M. Nucl. Instr. Meth., 1995, v. 106B, p. 298–301.
120. Weber W. J., Wang L. M., Yu N., Hess N. J. Mater. Sci. Engrg., 1998, v. 253A, p. 62–65.
121. Wendler E., Heft A., Wesch W. Nucl. Instr. Meth., 1998, v. 141B, p. 105–117.
122. Aihara J., Hojou K., Furuno S. et al. Nucl. Instr. Meth., 2000, v. 166/167B, p. 379–384.
123. Wang L. M., Weber W. J. Philos. Mag., 1999, v. 79, p. 237–241.
124. Weber W. J. J. Amer. Ceram. Soc., 1993, v. 76, p. 1729–1734.
125. Campisano S. U., Coffa S., Rainieri V. et al. Nucl. Instr. Meth., 1993, v. 80/81B, p. 514–517.
126. Bolse W. Nucl. Instr. Meth., 1998, v. 141B, p. 133–137.
127. Bolse W. Nucl. Instr. Meth., 1999, v. 148B, p. 83–88.
128. Lindner J. K.N. Nucl. Instr. Meth., 1996, v. 112B, p. 316–320.
129. Weber W. J. Nucl. Instr. Meth., 2000, v. 166/167B, p. 98–106.
130. Uzan-Saguy, C. Cyterman, Brener R. et al. Appl. Phys. Lett., 1995, v. 67, p. 1194–1196.
131. Prins J. F., Derry T. E. Nucl. Instr. Meth., 2000, v. 166/167B, p. 364–373.
132. Kalish R., Reznik A., Prawer S. et al. Phys. Stat. Sol. (a), 1999, v. 174, p. 83–99.
133. Jain U., Lidiard A. B. Philos. Mag., 1977, v. 35, p. 245–249.
134. Motta A. T., Olander D. R. Acta Mettal., 1990, v. 38, p. 2175–2181.
135. Pedraza D. F. J. Mater. Res., 1986, v. 1, p. 425–429.
136. Pedraza D. F. Radiat. Eff., 1990, v. 112, p. 11–17.
137. Lindner J. K.N. Nucl. Instr. Meth., 1992, v. 62B, p. 314–319.
138. Avrami M. J. Chem. Phys., 1941, v. 9, p. 177–182.
139. Damask A. C., Dienes G. J. Point Defects in Metals, Gordon and Breach, New York, 1963.
140. Weber W. J., Ewing R. C., Meldrum A. J. Nucl. Mater., 1997, v. 250, p. 247–151.
141. Weber W. J., Wang L. M., Yu N. Nucl. Instr. Meth., 1996, v. 116B, p. 322–326.
142. Weber W. J., Jiang W, Thevuthasan. Nucl. Instr. Meth., 2000, v. 166/167B, p. 410–414.
143. Weber W. J., Ewing R. C., Catlow C. R.A. et al. J. Mater. Res., 1998, v. 13, p. 1434–1438.
144. Roccaforte F., Gustafsson M. J., Bolse W. et al. Nucl. Instr. Meth., 2000, v. 166/167B, p. 148–153.
145. Roccaforte F., Bolse W., Lieb K. P. Nucl. Instr. Meth., 1999, v. 148B, p. 692–697.
146. Harbsmeier, Bolse W. J. Appl. Phys., 1998, v. 83, p. 4049–4053.
147. Dhar S., Bolse W., Lieb K. P. J. Appl. Phys., 1999, v. 85, p. 3120–3123.
148. Mc Hargue C. J., Farlow G. C., White C. W. et al. Mater. Sci. Eng., 1985, v. 69, p. 123–126.
149. Bigarrй J., Fayeulle S., Treheux D. J. Appl. Phys., 1997, v. 82, p. 3740–3746.
150. Abe H., Yamamoto Sh., Naramoto H. Nucl. Instr. Meth., 1997, v. 127/128, p. 170–175.
151. Mc Hargue C. J., Romana L. J. Nucl. Instr. Meth., 2000, v. 166/167B, p. 193–197.
152. Wang S. X., Wang L. M., Ewing R. S., Doremus R. H. J. Appl. Phys., 1997, v. 81, p. 587–593.
153. Shead L. L., Zinkle S. J., Hay J. C., Osborne M. C. Nucl. Instr. Meth., 1998, v. 141B, p. 123–132.
154. Heera V., Skorupa W., Pecz B., Dobos L. Appl. Phys. Lett., 2000, v.76, p..
155. Weselowski P., Lyutovich Y., Banhart F. et al. Appl. Phys. Lett., 1997, v. 71, p. 1948–1950.
156. Ewing R. S., Weber W. J., Lutze W., in: Disposal of Weapon Plutonium, eds: E. R.Merz, C. E.Walter, Kliower Academic Publishers, Boston, 1996, p. 65.
157. Wang S. X., Lumpkin G. R., Wang L. M., Ewing R. S. Nucl. Instr. Meth., 2000, v. 166/167B, p. 293–298.
158. Wang S. X., Wang L. M., Gong W. L., Ewing R. S. Nucl. Instr. Meth., 1998, v. 141B, p. 501–508.
159. Wang S. X., Wang L. M., Ewing R. S. Nucl. Instr. Meth., 1997, v. 127/128B, p. 186–190.
160. Wang S. X., Wang L. M., Ewing R. S. Nucl. Instr. Meth., 1998, v. 141B, p. 509–513.
161. Yu N., Sickafus K. E., Nastasi M. Philos. Mag. Lett., 1994, v. 70, p. 235–239.
162. Bordes N., Wang L. M., Ewing R. S., Sickafus K. E. J. Mater. Res., 1995, v. 10, p. 981–986.
Ион | Энергия КэВ | [dE/dx]n/ [dE/dx]el | Универсальный | Мольера | С-Kr | g | b | d,% | ||||||
Rp, нм | D Rp, нм | Nv | Rp, нм | D Rp, нм | Nv | Rp, нм | D Rp, нм | Nv | ||||||
1H+ | 100 | - | 1191 | 53,6 | 8,8 | 1192 | 53,6 | 8,6 | 1192 | 53,3 | 8,7 | -5,9 | 64,3 | - |
200 | - | 2321 | 60,1 | 9,8 | 2321 | 66,4 | 9,8 | 2321 | 59,8 | 9,8 | -6,5 | 78,1 | ||
300 | - | 3699 | 84 | 10,7 | 3699 | 83,1 | 10,7 | 3699 | 93,2 | 10,7 | -17 | 490,7 | ||
4He+ | 100 | - | 835 | 85,1 | 84,6 | 839 | 84,7 | 83,4 | 836 | 85,1 | 84,3 | -2,3 | 13,0 | |
200 | - | 1293 | 91,7 | 90,9 | 1292 | 95,9 | 91,1 | 1293 | 92,9 | 91 | -2,7 | 18,5 | ||
300 | - | 1656 | 94,7 | 94,5 | 1657 | 95,3 | 93,9 | 1655 | 96 | 94,6 | -2,8 | 20,1 | ||
10Ne+ | 50 | 0,84 | 119 | 29,5 | 313 | 117 | 28,3 | 317 | 119 | 29,0 | 315 | -0,64 | 3,3 | 1,6 |
100 | 0,46 | 238 | 48,9 | 485 | 237 | 47,7 | 491 | 238 | 48,5 | 488 | -0,93 | 4,1 | ||
200 | 0,25 | 471 | 76,4 | 698 | 471 | 76,5 | 701 | 471 | 76,9 | 703 | -1,35 | 5,8 | ||
40Ar+ | 50 | 1,83 | 67,5 | 16,6 | 382 | 63,3 | 15,7 | 398 | 66,5 | 16,3 | 387 | -0,28 | 2,8 | 6,2 |
100 | 1,16 | 129,3 | 28,8 | 642 | 124,2 | 27,8 | 664 | 128,1 | 28,8 | 648 | -0,44 | 2,9 | 3,9 | |
200 | 0,68 | 253,5 | 49,1 | 1022 | 249,2 | 48,0 | 1050 | 252,9 | 48,3 | 1033 | -0,66 | 3,4 | 1,7 | |
84Kr | 25 | 4,42 | 32,4 | 7,3 | 290 | 25,9 | 5,8 | 302 | 31,2 | 6,8 | 294 | 0,19 | 2,9 | 20 |
50 | 3,53 | 53,7 | 12,0 | 531 | 45,1 | 10,1 | 554 | 51,9 | 11,5 | 539 | 0,14 | 2,8 | 16 | |
100 | 2,80 | 93,4 | 20,5 | 944 | 82,3 | 18,5 | 981 | 91,2 | 19,7 | 959 | 0,09 | 2,8 | 12 | |
200 | 1,97 | 170,2 | 34,9 | 1624 | 157,5 | 33,6 | 1684 | 167,9 | 34,4 | 1650 | -0,05 | 2,6 | 7,5 | |
131Xe+ | 25 | 5,51 | 26,4 | 4,5 | 261 | 20,8 | 3,5 | 282 | 25,6 | 4,2 | 266 | 0,14 | 2,9 | 21,2 |
50 | 4,90 | 41,2 | 7,1 | 481 | 33,5 | 5,7 | 517 | 51,9 | 11,5 | 539 | 0,12 | 2,9 | 18,7 | |
100 | 4,10 | 66,3 | 11,2 | 858 | 56,2 | 9,7 | 918 | 64,4 | 10,7 | 875 | 0,03 | 2,8 | 15,2 | |
200 | 3,20 | 111,2 | 18,0 | 1470 | 98,2 | 16,4 | 1566 | 108,4 | 17,3 | 1503 | -0,07 | 2,8 | 11,7 | |
70Ga+ | 25 | 4,03 | 33,8 | 8,2 | 287 | 27,7 | 6,9 | 299 | 32,7 | 7,8 | 291 | 0,14 | 2,8 | 18 |
50 | 3,24 | 57,8 | 14,1 | 522 | 49,8 | 12,1 | 542 | 56,1 | 13,4 | 529 | 0,14 | 2,8 | 13,8 | |
100 | 2,38 | 103,5 | 24,5 | 921 | 93,4 | 22,3 | 954 | 101,3 | 23,6 | 935 | 0,04 | 2,7 | 9,8 | |
200 | 1,54 | 195,2 | 43,2 | 1573 | 182,8 | 41,1 | 1623 | 193,6 | 42,6 | 1599 | -0,11 | 2,7 | 6,3 | |
115In+ | 25 | 5,21 | 30,8 | 6,0 | 302 | 23,8 | 4,5 | 315 | 29,7 | 5,6 | 305 | 0,18 | 2,8 | 21,2 |
50 | 4,57 | 48,9 | 9,6 | 558 | 39,3 | 7,7 | 583 | 47,3 | 9,1 | 566 | 0,17 | 2,9 | 19,6 | |
100 | 3,75 | 85,1 | 16,1 | 1006 | 68,4 | 13,7 | 1049 | 78,8 | 15,2 | 1023 | 0,16 | 2,9 | 17,6 | |
200 | 2,84 | 142,4 | 27,6 | 1764 | 125,2 | 24,7 | 1834 | 139,2 | 26,6 | 1796 | 0,07 | 2,8 | 12,0 | |
122Sb+ | 25 | 5,31 | 30,7 | 5,8 | 303 | 23,8 | 4,4 | 316 | 29,7 | 5,5 | 306 | 0,20 | 3,0 | 22,4 |
50 | 4,68 | 48,7 | 9,3 | 562 | 38,8 | 7,5 | 586 | 46,2 | 8,7 | 570 | 0,15 | 2,8 | 20,3 | |
100 | 3,86 | 80,5 | 15,5 | 1015 | 67,1 | 13,0 | 1058 | 77,5 | 14,8 | 1030 | 0,15 | 2,9 | 16,6 | |
200 | 2,87 | 139,8 | 26,5 | 1783 | 121,6 | 23,5 | 1853 | 135,8 | 25,6 | 1814 | 0,08 | 2,8 | 13.0 |

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 |
