


Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
C. Отставание в психическом развитии, гипогликемия, ацетонурия, гиперлипидемия
D. Повышенная растяжимость кожи, разболтанность суставов, геморрагический диатез, плохое заживление ран
E. Аномалии скелета, глаз, сердечно-сосудистой системи
17.Какой тип кровоточивости (по Баркагану) характерен для синдрома Элерса-Данло:
A. Гематомний
B. Петехиально-пятнистый
C. Микроциркуляторо-гематомный
D. Васкулитно-пурпурный
E. Микроангиоматозный
18.Нарушение, какого звена гемостаза обуславливает появление геморрагий при синдроме Элерса-Данло:
A. Первичного
B. Вторичного
C. Третичного
D. Первичного и вторичного
19.К какой группе геморрагических заболеваний можно отнести геморрагии при синдроме Элерса-Данло:
A. Вазопатии
B. Тромбоцитопении и тромбоцитопатии
C. Коагулопатии
D. Смешаные (вазопатии и тромбоцитопатии)
20.Каков патогенез образования «рыбьего рта» при синдроме Элерса-Данло, из перечисленных ниже:
A. Является особенностью фенотипа синдрома Элерса-Данло
B. За счет атрофии мимических мышц
C. Вследствие постоянных микроразрывов соединительной ткани.
D. За счет гипертонуса мимических мышц
21.Какой из перечисленных ниже типов синдрома Элерса-Данло является наиболее тяжелым по клиническому течению:
A. Тип I
B. Тип II
C. Тип III
D. Тип IY
E. Cиндром Менкеса
22.Какой из перечисленных ниже типов синдрома Элерса-Данло наследуется сцеплено с Х-хромосомой:
A. Тип I
B. Тип II
C. Тип Y
D. Тип III
E. Тип IY
F. Cиндром Менкеса
23.Какой из перечисленных ниже типов синдрома Элерса-Данло наследуется аутосомно-рецессивно:
A. Тип II
B. Тип YIII
C. Тип Y
D. Тип III
E. Тип YI
F. Cиндром Менкеса
24.Из перечисленных ниже, выберите наиболее характерные клинические признаки VI типа синдрома Элерса-Данло:
A. Геморрагический синдром
B. Внутриглазничные кровоизлияния, отслойка сетчатки, глаукома
C. Гипопигментация кожи
D. Дегенеративные изменения нервной системы
E. Петлеобразная форма волос
25.Из перечисленных ниже, выберите наиболее характерные клинические признаки VIII типа синдрома Элерса-Данло:
A. Гипопигментация кожи
B. Гипремобильность суставов
C. Внутриглазничные кровоизлияния, отслойка сетчатки, глаукома
D. Геморрагический синдром
E. Дегенеративные изменения нервной системы
F. Периодонтит
26.Из перечисленных ниже, выберите наиболее характерные клинические признаки IX типа синдрома Элерса-Данло:
A. Периодонтит
B. Умственная отсталость
C. Дегенеративные изменения нервной системы
D. Гипопигментация кожи
E. Внутриглазничные кровоизлияния, отслойка сетчатки, глаукома
Раздел ІY -Б
СИНДРОМ МАРФАНА
Синдром Марфана - наследственное заболевание соединительной ткани с вовлечением скелетно-мышечной и сердечно-сосудистой систем и патологией глаз. Впервые описано Вильямсом в 1876 г.
Синдром Марфана (CM) - один из наиболее частых (5:100000) наследственных синдромов дизгистогенеза соединительной ткани. Тип наследования - аутосомно-доминантный, с высокой пенетрантностью гена. Частота встречаемости - 1 :
Синдром Марфана вследствие мутации гена фибриллина-1 (замена пролина на аргинин) локализованном в длинном плече 15 хромосомы, полеql5-q21.3).
В клинической картине со стороны скелетно-мышечной системы отмечается: высокий рост; астеническое телосложение; арахнодактилия (длинные тонкие пальцы); деформация грудной клетки; высокое арковидное нёбо; кифосколиоз; слабость связочного аппарата. Характерные изменения сердечно-сосудистой системы заключаются в дилатации корня аорты, аортальной регургитации, расслаивающейся аневризме аорты, пролапсе митрального клапана, регургитации крови при недостаточности митрального клапана. Имеет место патология глаз: иридоденез (дрожание хрусталика вследствие слабости цинновой связки), подвывих хрусталика, отслойка сетчатки, близорукость высокой степени. Редко встречаются геморрагический синдром и спонтанный пневмоторакс.
Диагностика
Для установления диагноза синдрома Марфана используют критерии нозологии Ghent, принятые в 1996 году, которые являются результатом пересмотра ранее существовавших критериев (Berlin) от 1988 года, которые представлены в таблице
Система | Главные критерии | Малые критерии |
Скелетная | Килевидная деформация грудной клетки; | Воронкообразная деформация грудной клетки; |
Воронкообразная деформация грудной клетки, требующая хирургического лечения; | Гипермобильность суставов; | |
Отношение длины верхнего сегмента тела к нижнему <0,86 или размаха рук к росту > 1,05; | Высокое небо и неровно растущие зубы; | |
Сколиоз (>20°) или спондилолистез; | Характерное лицо | |
Ограничение разгибания в локтевом суставе (<170°); | ||
Плоскостопие; | ||
Протрузия вертлужной впадины. | ||
Органы зрения | Вывих хрусталиков (эктопия зрения хрусталиков) | Увеличение аксиального размера глазного яблока (причина миопии); Уплощение роговицы; |
Гипоплазия радужки или целиарной мышцы (причина сужения зрачка). | ||
Сердечно-сосудистая | Дилатация корня аорты Расслоение восходящей аорты; | Пролапс митрального клапана; дилатация легочной артерии после сорокалетнего возраста; кальцификация митрального кольца после сорока лет; дилатация или расслоение иных участков аорты. |
Дыхатель-ная | Нет | Спонтанный пневмоторакс; |
Апикальные пузыри. | ||
Кожа | Нет | Атрофические стрии; Рецидивирующие грыжи |
Твердая мозговая оболочка | Эктазия пояснично-крестцового отдела. | Нет |
Генетичес-кие признаки | Наличие независимых критериев у родителей, детей или сибсов. Мутации, характерные для синдрома Мар-фана в гене фибриллина-1. Наследование маркерного гаплотипа ДНК, сцепленного с синдромом Марфана в семье. | Нет |
Таким образом:
· Для всех систем, кроме скелетной системы, достаточным условием считается наличие одного главного критерия или одного малого критерия.
· Для диагноза синдрома Марфана необходимо, как минимум, наличие по одному главному критерию в двух системах и одного малого в третьей. Изменение твердой мозговой оболочки и генетические признаки являются дополнительными критериями.
· Арахнодактилию выявляют с помощью признака большого пальца Steinberg (при сгибании большого пальца поперек ладони, его ногтевая фаланга выступает за ульнарный край) и признака Walker-Murdoch (перекрывание концевых фаланг большого пальца и мизинца при охвате ими запястья противоположной руки).
· Цифры, характеризующие соотношение сегментов тела, являются объективными критериями марфаноидного габитуса. Нижний сегмент измеряется от симфиза лобка до пола, длину верхнего сегмента получают вычитанием полученной цифры из роста пациента.
· Протрузию вертлужной впадины определяют путем рентгенографии тазобедренных суставов, эктазию твердой мозговой оболочки – МРТ исследованием.
· Дилатация аорты при синдроме Марфана, обычно, начинается с синуса Вальсальвы. Диаметр этого синуса, наряду с другими размерами аорты, измеряют при проведении трансторакальной ЭХО-КГ и оценивают в соответствии с возрастными нормами и площадью поверхности тела пациента. Увеличение диаметра корня аорты повышает риск ее расслоения. В качестве вспомогательных методов оценки состояния аорты, можно применять трансэзофагальную ЭХО-КГ, МРТ или КТ. Некоторые признаки синдрома Марфана, такие как эхокардиаграфические находки, эктопия хрусталика, сколиоз, соотношение верхнего сегмента к нижнему и протрузия вертлужной впадины, могут появиться по мере роста организма.
· Для молодых пациентов с семейной историей синдрома Марфана, которые не имеют убедительных клинических признаков, а также для молодых пациентов с Марфаноподобными признаками, но без семейного анамнеза, необходимо постоянное наблюдение до возраста 18 лет.
· Пробанд с подозрением на синдром Марфана должен наблюдаться клиническим генетиком, кардиологом, офтальмологом и врачом лучевой диагностики, в соответствии со следующим алгоритмом.
Пациентка М, 17 лет. Диагноз синдром Марфана, поставлен в 3 года.
При обследовании выявлено: миопия высокой степени, дислокация хрусталиков, сколиоз, дилатация корня аорты с недостаточностью клапана, пролапс митрального клапана с его недостаточностью.
Фото 1 Сколиоз. Обратите внимание на рост пациентки по сравнению с ее матерью.
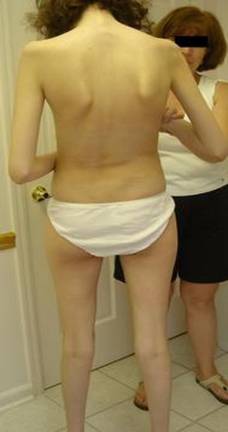
Фото 2 Деформация грудной клетки

Фото 3 Арахнодактилия пальцев стопы

Фото 4 Арахнодактилия пальцев кисти

Дифференциальный диагноз. Существует много заболеваний, имеющих клиническую картину аналогичную синдрому Марфана.
Так, например гомоцистинурия, для которой характерны нарушения строения скелетно-мышечной и сердечно-сосудистой систем. Однако, при гомоцистинурии выявляется умственная отсталость, а при анализе мочи – гиперэкскреция гомоцистина и метионина, что не определяется при синдроме Марфана.
Синдром Билса (Beals syndrome или врожденной контрактурной арахнодактилии, ассоциированной с мутациями в гене фибриллина 2). Является разновидностью болезни Марфана с нарушением синтеза белка фибриллина-2, что проявляется контрактурами суставов и аномалией строения ушной раковины.
Трисомия 8 имеет костные проявления, как при синдроме Марфана, но без глазных и сердечно-сосудистых признаков. Марфаноидное телосложение с микроцефалией и гломерулонефритом. Лабораторные исследования неспецифичны. Отмечают непостоянную гиперэкскрецию гликозаминогликанов и оксипролина с мочой.
Марфаноидный фенотип встречается при синдроме Lujan-Fryns (Х-сцепленное заболевание), синдроме Shprintzen-Goldberg (аутосомно-доминантное заболевание) и краниосиностозе. У некоторых больных с синдромом Shprintzen - Goldberg обнаружены мутации в гене фибриллина 1, что может быть расценено как необычный вариант синдрома Марфана.
Другие болезни отличить от синдрома Марфана бывает сложно. Например, синдром MASS (миопия, пролапс митрального клапана, дилатация аорты, изменение кожи и скелета) можно расценить, как умерено выраженный синдром Марфана. При этом заболевании иногда находят мутации в гене фибриллина-1, однако дилатация аорты у больных менее выражена, и риск развития аневризмы низкий. Кроме того, встречается сочетание пролапса митрального клапана с деформацией грудной клетки (чаще воронкообразной), но без других признаков синдрома Марфана. В одном из таких случаев была обнаружена мутация в гене фибриллина 1, однако широких молекулярно-генетических исследований этого заболевания не проводилось. Изменения скелета, характерные для синдрома Марфана и мутации в гене фибриллина 1 описаны при аутосомно-доминантной эктопии хрусталиков. Описана также изолированная наследственная аневризма аорты, ассоциированная с еще некартированными генами на 5 и 11 хромосоме.
Если ни один из перечисленных диагнозов не поставлен, у больного - неклассифицированный комплекс дефектов развития и марфаноподобный фенотип (арахнодактилия плюс долихостеномелия).
Лечение. Приемлемым для пациентов с синдромом Марфана является низкий или средний уровень физической активности. Из-за риска сердечно-сосудистых осложнений и возможной дислокации хрусталиков, не рекомендуется заниматься контактными видами спорта, при которых существует вероятность телесного повреждения (например, регби, прыжки, конный спорт). Следует также избегать подводного плавания, из-за риска развития пневмоторакса.
Медикаментозное лечение больных с синдромом Марфана заключается в коллагеннормализующая терапия. Метод основан на коррекции метаболизма коллагена и других компонентов соединительной ткани.
Симптоматическое лечение проявлений синдрома Марфана касается коррекции зрения и поражений скелета. Офтальмологические нарушения (миопия, астигматизм) подлежат коррекции с помощью очков либо контактных линз, устранению подлежат в первую очередь воронкообразная и килевидная деформация грудной клетки.
Наиболее частым поражением скелета является воронкообразная деформация грудной клетки (ВДГК). В основе ВДГК лежит дизгистогенез реберных хрящей при опережении их роста; выраженность деформации определяют с помощью индекса Гижицкой. Лечение ВДГК только хирургическое – торакопластика (попытки консервативной профилактики и лечения к успеху не приводят). Показания к торакопластике функциональные. При западении передней стенки грудной клетки происходит снижение объема грудной полости и недостаточное расправление легких. Это приводит к уменьшению площади альвеолярных мембран, снижению диффузии кислорода из альвеолярного воздуха в кровь. Организм компенсирует это состояние усилением легочного кровотока (возникает перегрузка правых отделов сердца), снижением потребления кислорода за счет уменьшения массы тела и увеличением кислородной емкости крови. Торакопластика показана при воронкообразной деформации грудной клетки II-III степени, а также если выявляются два из трех признаков перенапряжения кардиореспираторной системы: ЭКГ-признаки перегрузки правых отделов сердца; дефицит массы тела более чем на 20% от среднеевропейской нормы; увеличение числа эритроцитов крови, уровня гемоглобина и (или) цветового показателя более чем на 20% от нормы. Оперировать больных следует в возрасте старше 4 лет; оптимальный возраст для операции - 5-7 лет. Часто возникающие у больных этой группы интра - и послеоперационные осложнения обусловливают высокую послеоперационную летальность (до 30%). В настоящее время операции осуществляются по методике Саламаа-Палтиа в собственной модификации. К операции по поводу килевидной деформации грудной клетки при синдроме Марфана следует подходить еще осторожнее (килевидная деформация не вызывает функциональных нарушений, и показания к ее хирургической коррекции исключительно косметические). Рекомендуется методика Равича на фоне коллагеннормализующей терапии.
Сердечно-сосудистые осложнения включают: пролапс и регургитацию митрального клапана, дилатацию левого желудочка и легочной артерии. Но наиболее общей причиной ухудшения состояния и смертности является дилатация корня аорты. Несостоятельность аортального клапана, обычно, как следствие дилатации корня аорты, и риск разрыва аорты значительно повышается, если диаметр синуса Вальсальвы превышает 5,5 см у взрослых. Окклюзия коронарных стволов расслаивающимся корнем аорты может привести к инфаркту миокарда. Аорта больных с синдромом Марфана гистологически характеризуется фрагментацией и беспорядочным расположением эластичных фибрилл, особенно в гладкомышечных клетках. Коллаген и мукополисахариды, накапливающиеся между клетками, создают картину "кистозной медиальной дегенерации", хотя истинные кисты отсутствуют. Мукополисахариды, откладывающиеся в клапанах, могут стать причиной утолщения их стенок. Молекулярной основой этих изменений является аномальный фибриллин – важный компонент миофибрилл. Дегенерация фибрилл аорты приводит к снижению эластичности аорты и увеличению скорости пульсовой волны. Это может быть подтверждено ЭХО-КГ в любом возрасте, или МРТ, хотя у детей она менее показательна. В норме, с возрастом, аорта постепенно расширяется и становится менее упругой, но при синдроме Марфана возрастные изменения более заметны.
Больным с синдромом Марфана назначают пропранолол, атенолол и метопролол, которые увеличивают эластичность и снижают резистентность аорты, а также скорость пульсовой волны. Абсолютным показанием для назначения бета-блокаторов является дилатация аорты более 4 см.
Выделены факторы риска расслоения аорты при синдроме Марфана:
· диаметр аорты > 5 см у взрослых;
· распространение дилатации за пределы синуса Вальсальвы;
· быстро прогрессирующая дилатация (> 5% или 2 мм в год, у взрослых); семейные случаи расслоения аорты
Все пациенты с синдромом Марфана ежегодно должны проходить клинический осмотр и трансторакальную ЭХО-КГ. У детей ЭХО-КГ рекомендуется проводить с интервалом 6-12 месяцев, в зависимости от диаметра и скорости дилатации аорты. Профилактическое хирургическое вмешательство на аорте должно быть предпринято, если диаметр синуса Вальсальвы превышает 5,5 см у взрослых и 5,0 см у детей.
Предлагается использовать антагонисты кальция или ингибиторы ангиотензин конвертирующего фермента, хотя пока это лишь теоретические предположения. На культуре клеток аорты больных с синдромом Марфана показано, что ингибиторы ангиотензин конвертирующего фермента и блокаторы рецепторов ангиотензина II препятствуют апоптозу гладкомышечных клеток сосудов.
Тест-контроль к разделу ІY -Б
27.Синдром Марфана – наследственное заболевание соединительной ткани с вовлечением, перечисленных ниже структур:
A. Кроветворной системы
B. Скелетно-мышечной и сердечно-сосудистой систем, патологией глаз
C. Эндокринной системы
D. Связочного аппарата, сухожилий
E. Центральной нервной системы
28.Какой тип наследования синдрома Марфана:
A. Аутосомно-доминантный
B. Аутосомно-рецессивный
C. Рецессивный, сцепленный с Х-хромосомой
D. Неизвестный
29.Клиническая картина синдрома Марфана обусловлена:
A. Недостаточностью коллагена
B. Мутацией в гене фибриллина-1
C. Высоким уровнем лейцина
D. Высоким уровнем гомоцистина
30.Для синдрома Марфана характерна перечисленная ниже симптоматика:
A. Гиперакузия, плохой сон, задержка двигательного развития, симптом «вишневой косточки» на глазном дне
B. Аномалии скелета, ломкость и хрупкость костей, глухота
C. Отставание в психическом развитии, гипогликемия, ацетонурия, гиперлипидемия
D. Высокий рост, астеническое телосложение, арахнодактилия, деформация грудной клетки, арковидное нёбо
E. Повышенная растяжимость кожи, разболтанность суставов, геморрагический диатез, плохое заживление ран
31.Для синдрома Марфана наиболее характерными являются перечисленные ниже изменения со стороны сердечно-сосудситой системы:
A. Развитие синдрома Рейно
B. Врожденные пороки сердца (тетрада Фалло, дефект межжелудочковой перегородки, транспозиция магистральных сосудов)
C. Дилатация корня аорты, аортальная регургитация, расслаивающаяся аневризма аорты, пролапс митрального клапана
D. Формирование гипретонической болезни
32.Из перечисленных ниже видов спорта выберите рекомендуемые, для детей с синдромом Марфана:
A. Бокс, регби
B. Ходьба на лыжах, плавание
C. Прыжки
D. Конный спорт
E. Дайвинг
F. Парашютный спорт
33.Патогенетическое лечение детей с синдромом Марфана включает в себя:
A. Гемостатическую терапию
B. Коллагеннормализующее лечение
C. Коррекцию аномалий развития скелета
D. Аортопластику
34.Симптоматическое лечение детей с синдромом Марфана включает в себя:
A. Гемостатическую терапию
B. Коррекцию зрения и поражений скелета, аортопластику
C. Иммунокоррегирующую терапию
D. Витаминотерапию
35.Какие наиболее частые осложнения со стороны сердечно-сосудистой системы при деформации грудной клетки у детей с синдромом Марфана:
A. Приобретенные пороки сердца
B. Гипертоническая болезнь
C. Артериальная гипотензия
D. Кардиомиопатия
E. Врожденные пороки сердца
36.Показаниями для торакопластики у детей с синдромом Марфана являются:
A. Воронкообразная деформация грудной клетки II-III степени
B. Частые острые респираторные заболевания
C. Перегрузка левых отделов сердца
D. Возраст до 4-х лет
37.Препаратами выбора для профилактики дилатации и расслоения аорты являются:
A. Бета-блокаторы
B. Витамины
C. Препараты L-карнитина
D. Блокаторы кальциевых каналов
38.Факторами риска расслоения аорты при синдроме Марфана являются:
A. Воронкообразная деформация грудной клетки II-III степени
B. Диаметр аорты >5 см, распространение дилатации за пределы синуса Вальсальвы, быстро прогрессирующая дилатация (> 5% или 2 мм в год, у взрослых) семейные случаи расслоения аорты
C. Диаметр аорты ≤ 5 см, прогрессирующая дилатация > 5% в год, у взрослых, семейные случаи расслоения аорты
D. Перегрузка левых отделов сердца
E. Возраст до 4-х лет
39.Как часто, у детей с синдромом Марфана, рекомендуется проводить ЭХО-КГ:
A. Один раз в 6-12 месяцев
B. Один раз в месяц
C. Один раз в 3 года
D. Один раз в квартал
Раздел ІY -В
НЕСОВЕРШЕННЫЙ ОСТЕОГЕНЕЗ
Несовершенный остеогенез (болезнь Лобштейна) — наследственное заболевание, характеризующееся генерализованным дефицитом костной массы (остеопения) и чрезвычайно выраженной хрупкостью, ломкостью костей. Приблизительно у 95% больных имеются мутации генов, кодирующих синтез про-а, и про-а2 пептидов коллагена 1-го типа.
Частота заболевания различна в зависимости от типа. І тип несовершенного остеогенеза встречается с частотой 1 на, II тип — с частотой 1 на, III и IV типы — с частотой 1 начеловек.
Наиболее признанной классификацией несовершенного остеогенеза является классификация, предложенная Sillence. Выделяют 4 типа заболевания в зависимости от клинических проявлений и типа наследования.
Классификация несовершенного остеогенеза (по Sillence)
Тип | Ломкость костей | Голубые склеры | Аномалии зубов | Глухота | Тип наследования |
I | Легкая степень | Определяются | Отсутствуют при подтипе І А, имеются при подтипе І Б | Имеется у большинства больных | Аутосомно - доминантный |
II | Резко выраженная | Определяются | Имеются в некоторых случаях | Отсутствует | Спорадический, реже - аутосомно- рецессивный |
III | Выраженная | Голубой цвет склер при рождении | Имеются в некоторых случаях | Встречается очень часто | Аутосомно - рецессивный |
IV | Вариабельный признак | Не опреде - ляются | Отсутствуют при ІVА подтипе выявляются При ІVБ подтипе | Встречается очень часто | Аутосомно - доминантный |
Клиническая картина

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 |
