
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
На основании полученных кинетических данных были проведены расчеты образующейся топологической структуры. Получены качественные и количественные расчетные характеристики топологической структуры полимерных матриц, которые коррелируют с экспериментальными данными: конверсия функциональных групп, золь и гель фракции.
оценка топологической структуры трехкомпонентных эпоксиАминных матриц, получаемых в естественных условиях.
, , *
Казанский государственный технологический университет,
Казань, Россия, E-mail: *****@***ru
*Институт проблем химической физики РАН,
Черноголовка, Россия
В ранее представленных работах было создано описание формирования топологической структуры эпоксиаминной матрицы. Описание основано на концепции блоков связей, которая сочетает в себе статистический и кинетический подходы, с учетом двух упрощений Флори: реакционная способность молекул не зависит от их размера и не происходит реакций циклизации. Использование концепции позволяет спрогнозировать топологическую структуру формирующейся эпоксиаминной матрицы, показать распределение узлов сетки по функциональность, определить критическую конверсию, величины золь и гель фракции. Сравнение модельных систем показало адекватность модели реально протекающему процессу.
Как продолжение, описание было расширено, тем самым позволив рассматривать трехкомпонентные системы, в которых третьим компонентом является эпоксидный модификатор, причем функциональность модификатора может варьироваться.
Используя полученные кинетические данные были рассчитаны топологические структуры эпоксиаминных матриц, содержащих моно - и бифункциональные эпоксидные модификаторы различного строения.
Были получены топологические картины, исходя из которых были сравнены расчетные значения критической конверсии и золь-гель фракции с экспериментальными данными. Сравнение показало, как и в случае модельных систем (не содержащих модифицирующие добавки), адекватность представленного описания.
РЕАКЦИОННАЯ СПОСОБНОСТЬ СИЛИЛЬНЫХ, ГЕРМАНИЛЬНЫХ И СТАННИЛЬНЫХ РАДИКАЛОВ В РЕАКЦИЯХ ПРИСОЕДИНЕНИЯ
Институт проблем химической физики РАН,
Черноголовка, Россия, e-mail: *****@
Разработана новая полуэмпирическая модель реакций радикального присоединения. Модель рассматривает переходное состояние реакции X· + Y=Z ® XYZ· как результат суперпозиции трех потенциальных кривых, каждая их которых описывает потенциальную энергию валентного колебания связи Y=Z, Y-Z и X-Y как параболическую функцию амплитуды ее колебания. Реакция присоединения характеризуется энтальпией DH, энергией активации Е, коэффициентами a, g и b, каждый из которых – сочетание силовых постоянных реагирующих связей, и параметром re – алгебраической суммой удлинения связей в переходном состоянии. Каждый класс реакций характеризуется классическим потенциальным барьером термонейтральной реакции: Ee01/2 = bre/(a+g). С использованием этих уравнений проанализировано 37 классов реакций присоединения.
Анализ полученных значений Ee0 показал, что большое влияние на энергию активации оказывает триплетное отталкивание в переходном состоянии. Проявляется это в том, что Ee0 ~ re2, а зависимость параметра re от De(X-Y) и от радиуса атома, несущего свободную валентность и атакующего двойную С=С-связь, имеет вид: re /De(X-C)/моль м кДж-1 = 5.3 ´ 10-13 + 2.4 ´ 10-9(De ´ rC-X)1/2 , где X = Si, Ge, Sn, а rC-X - длина X-C-связи. Это уравнение отражает тот факт, что радиус атома, атакующего двойную связь, влияет на энергию активации, но это влияние, в свою очередь, зависит от триплетного отталкивания. В реакциях силильных радикалов с симметричными производными этилена проявляет себя стерический эффект: Ee0(CH2=CHR) < Ee0(RCH=CHR) и разность DES = Ee0(RCH=CHR) - Ee0(CH2=CHR) = 27.2 кДж/моль.
В реакциях Et3Si·, Bu3Ge· и Bu3Sn· с диенами и стиролами проявляется взаимодействие электронов переходного состояния с
p-электронами соседней С=С-связи или фенильного кольца, повышающее энергию активации присоединения на 22-39 кДж/моль.
ПОЛЯРНЫЙ ЭФФЕКТ В РЕАКЦИЯХ ПОЛИМЕРИЗАЦИИ И СОПОЛИМЕРИЗАЦИИ МОНОМЕРОВ
Институт проблем химической физики РАН,
Черноголовка, Россия, e-mail: *****@
В рамках модели реакции радикального присоединения как суперпозиции трех потенциальных кривых, описывающих потенциальную энергию валентного колебания связи как функцию амплитуды ее колебания, проанализированы экспериментальные данные (константы скорости) радикальной полимеризации и сополимеризации полярных мономеров. Такой подход позволяет по экспериментальным данным вычислить энергию активации термонейтрального аналога рассматриваемой реакции и разделить вклад энтальпии реакции, триплетного отталкивания и полярного взаимодействия в энергию активации присоединения мономера к растущему макрорадикалу.
Ниже приводится пример такой оценки для реакций полимеризации ряда полярных мономеров. В качестве реакции сравнения взята реакция продолжения цепи при полимеризации этилена. Вклад энтальпии в энергию активации вычислялся как разность:
DEH = Е – Е0. Вклад взаимодействия полярных групп вычислялся как разность между энергиями термонейтральных реакций присоединения полярного мономера и этилена: DEm = E0 (CH2=CHX) – E0(CH2=CH2).
Полимер | –DH | E | E0 | DEH | DEm |
кДж/моль | |||||
CH2=CH2 | 93.5 | 37.4 | 119.2 | –81.8 | 0.0 |
CH2=CHOAc | 114.9 | 30.4 | 118.4 | –88.0 | –0.8 |
CH2=CHCO2Me | 96.3 | 31.0 | 114.5 | –83.5 | –4.7 |
CH2=CMeCO2Me | 98.5 | 33.0 | 108.1 | –75.1 | –11.0 |
CH2=CHCN | 119.7 | 20.2 | 111.0 | –90.8 | –8.2 |
CH2=CHCl | 120.7 | 24.0 | 116.0 | –92.0 | –3.2 |
Как видно из приведенных данных, полярный эффект снижает энергию активации и в ряде случаев весьма значительно (на 8 ¸ 11 кДж/моль). После энтальпии реакции это самый существенный фактор, влияющий на энергию активации индивидуальной реакции.
КОМБИНАТОРНАЯ СИММЕТРИЯ ТОПОЛОГИЧЕСКИХ СВЯЗЕЙ И ЭФФЕКТ СЛИЯНИЯ ЗВЕНЬЕВ И МОД ПРИ ВЯЗКОУПРУГОЙ РЕЛАКСАЦИИ РАЗВЕТВЛЕННЫХ ОЛИГОМЕРОВ
,
ИПХФ РАН, г. Черноголовка, Россия, *****@
Рассматривается линейная модель релаксации, в которой макромолекулы олигомеров представляются как система легких упруго связанных частиц, движущихся в вязкой среде. Нередко в структуре графа топологических связей разветвленных макромолекул присутствует симметрия относительно перестановок. Например, если переставить и перенумеровать от "корня" две ветви правильной звездообразной макромолекулы, то вид уравнений движения сохранится. Такая же ситуация имеет место для сверхразветвленной макромолекулы с бинарным графом связей: можно переставлять ветви, исходящие из общего узла и при этом уравнения движения останутся неизменны.
Наличие единой перестановочной симметрии в топологических связях, модулях упругости и коэффициентах трения и начальных данных приводит к тому, что группы узлов и связей "сливаются" и движутся как единое целое. Это вытекает из следующего: если коэффициенты трения и упругой связи, начальные условия звеньев инвариантны относительно некой перестановки - симметрии, то и компоненты решения уравнений релаксационного движения инвариантны относительно этой перестановки. Из этого простого предложения вытекает, что звенья попадающие в непересекающиеся циклические классы, порожденные перестановкой-симметрией совпадают в ходе релаксационного движения.
В итоге движение исходной макромолекулы точно описывается сокращенной агрегированной системой уравнений движения, которая получается суммированием параметров трения, связи слившихся звеньев. Поскольку число релаксационных мод в разложении целевой функции-модуля (например, напряжения) связано с размерностью системы уравнений движения, из-за эффекта слияния косвенно сокращается и число ненулевых компонент экспоненциального разложения, т. е. релаксационный спектр. В докладе показано, что следствием эффекта слияния является эквивалентность релаксации свернутых звезд и гиперразветвленных молекул релаксации неоднородных линейных цепей, размер которых не превосходит удвоенной длины ветвей и числа поколений, причем явно указаны параметры этих цепей.
Работа выполнена при поддержке РФФИ, проекты , .
КРИТИЧЕСКАЯ КОНВЕРСИЯ ПРИ ЦИКЛОТРИМЕРИЗАЦИИ.
,
Институт проблем химической физики РАН
Черноголовка, Россия, irzhak@icp.ac.ru.
Реакция циклотримеризации, которая характерна для изоцианатов, производных циановой кислоты и алкинов, с кинетической точки зрения представляет собой особый случай поликонденсационных процессов, ибо рост полимерной цепи осуществляется за счет взаимодействия не двух, как обычно, а трех функциональных групп. Вне зависимости от химических особенностей механизма реакции ее можно представить как тримолекулярную в некотором фиктивном времени.
Предложена общая схема циклотримеризации моно - и бифункциональных олигомеров без учета эффекта замещения, но с учетом разной реакционной способности функциональных групп с допущением аддитивности свободной энергии активации, т. е. в предположении, что константу скорости реакции А+В+С kabc можно выразить как kabc=kakbkc. Приведены результаты расчетов для зависимости величины критической конверсии aС от состава реакционной смеси и относительной реакционной способности функциональных групп. Обнаружен экзотический характер зависимости, а именно, уменьшение величины критической конверсии от концентрации обрывателя цепи развития сетки, монофункционального агента, в случае его низкой реакционной способности, обусловленный тем, что сетка формируется, главным образом, за счет бифункционального агента, и aС падает из-за снижения его доли.
Учет эффекта замещения ведет к существенному усложнению схемы реакции вследствие появления дополнительной константы скорости k3, характеризующей реакционную способность второй функциональной группы В®C, если первая прореагировала. В связи с этим приходится различать не четыре типа реакций, как выше, а десять.
Приведены результаты расчетов, показывающих зависимость величины критической конверсии aС от состава реакционной смеси и величины эффекта замещения, т. е. относительной реакционной способности функциональных групп С. Показано, что положительный эффект замещения при относительно невысоком содержании монофункционального компонента понижает величину критической конверсии, тогда как отрицательный ведет к некоторому повышению aС.
ПОЛЯРНЫЕ ЭФФЕКТЫ В РЕАКЦИЯХ ПРИСОЕДИНЕНИЯ ФЕНОКСИЛЬНЫХ И НИТРОКСИЛЬНЫХ РАДИКАЛОВ К ДВОЙНЫМ СВЯЗЯМ ОЛИГОМЕРНЫХ ЭФИРОВ (МЕТ)АКРИЛОВОЙ КИСЛОТЫ
, ,
Ярославский государственный университет им. ,
г. Ярославль, Россия, pliss@uniyar.ac.ru
Изучена кинетика присоединения 2,4,6-три(трет. бутил)феноксиль-ного (PhO.) и 4,4-диметоксидифенилнитроксильного ([>NO.) радикалов к p-связям олигоэфиров (мет)акриловой кислоты и полиолов (метод ЭПР). Расчет констант скорости по экспериментальным данным проводили по оригинальной оптимизационной программе. Расчет энтальпии присоединения радикалов осуществлялся квантовохимически (методом АМ1).
Изменение концентрации PhO· { Ar, ([PhO·]=1моль/л, [>C=C<]=0.5-4,5 моль/л)} хорошо описывается зависимостью kPhO·t=ln([PhO·]0/[PhO·])/ Кинетика расходования >NO×) в принятых экспериментальных условиях {(Ar, [>NO·]= моль/л, [![]() ]=0.5-4,5 моль/л} также описывается уравнением: ln([>NO·]0/[>NO·])=k NO· t.
]=0.5-4,5 моль/л} также описывается уравнением: ln([>NO·]0/[>NO·])=k NO· t.
Полученные данные (Таблица) свидетельствуют, что по мере увеличения числа (мет)акрилатных групп в олигомере значения kPhO· и kNO· (в расчете на одну p-связь)существенно падают по сравнению с моноэфирами (бутилакрилат и метилметакрилат).
105kPhO· и108 (kNO·, л/( моль.с), 353К | |||||||
Олиго-мер | БА (1) | НГДА (2) | ТПТА (3) | ПЭТА (4) | ММА (1) | ТПТМ (3) | ПЭТМ (4) |
PhO. | 5,.1 | 4.8 | 0,7 | 0,6 | 1,1 | 0,4 | 0,3 |
>NO· | 2,2× | 0,3 | 0,2 |
В скобках – число (мет)акрилатных групп в олигоэфире
Данная феноменология обусловлена эффектом мультидипольного взаимодействия (), обнаруженным ранее для присоединения RO2×, ROOH и O2 к p-связям данных олигоэфиров (, с сотр.). Существенная разница в значениях kPhO· и kNO· (три порядка) обусловлена различиями в термохимии присоединения PhO· и >NO..
ВНУТРИМОЛЕКУЛЯРНАЯ ПОДВИЖНОСТЬ ЦЕПЕЙ
ПОЛИ(ФТОР)АЛКИЛМЕТАКРИЛАТОВ
, ,
Ярославский государственный технический университет,
г. Ярославль, *****@***ru
В последнее время установлена чувствительность кинетики полимеризации высших алкил(мет)акрилатов к введению малых добавок (1-5%) других алкил(мет)акрилатов [, , Могилевич М. М., , // Высокомолек. соед.- А.- 2003. Т. 45. № 6. С. 892.] с той же реакционноспособностью. Так как мономер-добавка расходуется в процессе полимеризации как и основной мономер, встраиваясь в растущую полимерную цепь, то эти кинетические эффекты, вероятно, должны приводить к структурным изменениям синтезируемого полимера и в конечном счете сказываться на его свойствах.
Цель данной работы – моделирование внутримолекулярной подвижности полимерных цепей полидодецилметакрилата (ПДДМА), получаемых в отсутствии и присутствии 4% мол. бутилметакрилата (БМА), а также полимерных цепей полидодекафторгептилметакрилата (ПДФГМ), получаемых в отсутствии и присутствии 4% мол. тетрафторпропилметакрилата (ТФПМ).
Объектами для компьютерного моделирования служили 50-звенные фрагменты полимерных цепей ПДДМА (ПДФГМ) и ПДДМА, (ПДФГМ), у которых 17 и 34 звеньями являются звенья БМА (ТФПМ). Для моделирования внутримолекулярной подвижности фрагментов полимерных цепей использовался метод молекулярной динамики – проводилось численное интегрирование уравнений движения Ньютона с использованием алгоритма Беемана [ Методы компьютерного эксперимента в теоретической физике. – М: Наука, 1990. – 176 с.].
Статистический анализ динамического поведения фрагментов полимерных цепей ПДДМА (ПДФГМ) и ПДДМА-БМА (ПДФГМ-ТФПМ) в течение 1000 пс после достижения «тепловой релаксации» при заданных температурах (в диапазоне 50 – 300 К) показал, что замена двух ДДМА (или ДФГМ) звеньев в 50-звенных полимерных фрагментах ПДДМА (ПДФГМ) на два звена БМА (ТФПМ) приводит к значительному увеличению флуктуации средних расстояний между конечными атомами углерода основных полимерных цепей и косинусов двухгранных углов в местах локализации инородных звеньев.
МАТЕМАТИЧЕСКОЕ МОДЕЛИРОВАНИЕ КИНЕТИКИ РАДИКАЛЬНО-ЦЕПНОЙ ПОЛИМЕРИЗАЦИИ С УЧЕТОМ АССОЦИАЦИИ РЕАГЕНТОВ
, ,
Институт проблем химической физики РАН,
Черноголовка, Россия, *****@
Олигомеры, а также мономеры, образуют ассоциаты (домены, кластеры и т. д.), причем реакционная способность последних, как правило, заметно отличается от таковой для исходных реагентов [, , . Олигомерное состояние вещества М.; Наука. 2005]. Между тем, надмолекулярная структура реагентов обычно не принимается во внимание при расчете кинетических параметров по экспериментальным данным о радикально-цепной полимеризации, и, таким образом, расчетные константы скорости являются по существу эффективными характеристиками, зависящими как от истинной константы скорости полимеризации, так и от константы равновесия процесса ассоциации. Это обстоятельство не дает возможность выявить детальный механизм и определить истинные кинетические характеристики радикально-цепной полимеризации в жидкой фазе. Это, в свою очередь, в какой-то степени препятствует развитию теоретических основ управления кинетикой этих реакций за счет внешних физических воздействий. Так, в частности, ни одна из теоретических моделей, описывающих влияние звука на скорость химической реакции, не учитывает изменение надмолекулярной структуры реагентов под воздействием звука [. Звукохимические реакции и сонолюминесценция. М: Химия. 1986].
В работе предложена новая математическая модель кинетики радикально-цепной полимеризации, включающая, наряду со стадиями инициирования (с образованием радикальной пары и выхода радикалов из клетки), роста и обрыва кинетической цепи, стадий образования и распада ассоциатов реагентов. Анализ системы кинетических уравнений проводился численным методом. В приближении квазистационарности концентрации радикалов в аналитическом виде получены соотношения, связывающие скорость полимеризации с константой равновесия ассоциации реагентов. Исследовано влияние напряжений, создаваемых акустическим воздействием, на скорости стадий распада ассоциатов, образования радикальной пары и рекомбинацию радикалов. Показано, что скорость процесса полимеризации определяется в основном величиной амплитуды акустических колебаний.
Синтез олигомеров.
Новые функциональные олигомеры. Получение и свойства
МЕХАНИЗМ ОКИСЛЕНИЯ ОЛИГОМЕРОВ ДЕЦЕНА-1
, ,
Институт проблем химической физики РАН,
г. Черноголовка, Россия. E-mail: *****@
На основе разработанного в ИПХФ РАН метода катионной олигомеризации олефинов создана технология и построен завод по производству синтетических олигодеценовых масел. С целью повышения окислительной стабильности производимых масел ведется работа по исследованию процесса окисления основных продуктов олигомеризации децена-1. В настоящей работе проведено кинетическое исследование механизма окисления фракции ПАО-2, более чем на 90% состоящей из димеров децена-1. При 120, 130 и 140°С изучены кинетические закономерности автоокисления и инициированного окисления исходного и гидрированного образцов ПАО-2, идентифицированы ключевые реакции в механизме процесса и определены численные значения кинетических параметров. Установлено, что причиной высокой окисляемости исходного олигомера, кроме наличия в образце двойных связей и гидропероксидов, являются большие значения скорости самопроизвольного зарождения радикалов и константы скорости распада гидропероксидов на радикалы, что связано с наличием в исследуемом образце различных примесей, в том числе каталитического характера.
Показано, что в результате гидрирования исходного ПАО-2 происходит положительное изменение всех параметров окисления, что и приводит к увеличению окислительной стабильности олигомера.
Проведено сравнительное исследование механизма окисления образца ПАО-2, полученного по измененной технологии. Показано, что от технологии производства зависят как качественные, так и количественные показатели окислительной стабильности получаемого олигомера. Внесены необходимые дополнения в механизм окисления нового образца и определены значения соответствующих параметров.
С целью повышения окислительной стабильности исходных, негидрированных продуктов олигомеризации проведено исследование процесса окисления образца ПАО-2 в присутствии антиоксидантов класса пространственно-затрудненных фенолов и ароматических аминов. Показано, что кинетические закономерности ингибированного окисления ПАО-2 соответствуют классическим представлениям теории жидкофазного окисления, и рассмотренные ингибиторы могут быть использованы для защиты от окисления первичных продуктов олигомеризации в период их временного хранения.
ОЛИГОДИЕНЫ С КОНЦЕВЫМИ ФУНКЦИОНАЛЬНЫМИ ГРУППАМИ. СТРОЕНИЕ И СВОЙСТВА.
, ,
Институт химии высокомолекулярных соединений НАН Украины
Киев, Украина, E-mail: *****@***net
Изучены закономерности синтеза и свойства олигодиенов с концевыми функциональными группами: гидразидными, ацилгидразонными, уретанизоцианатными, оксадиазолинил-карбаматизоцианатными.
Показано влияние образования водородных связей между концевыми функциональными олигодиенов группами на их реологические характеристики, а также на физико-механические свойства полученных на их основе полимерных материалов.
При сравнении строения и реологических характеристик функциональных олигодиенов установлено: чем выше уровень водородных связей между полярными группами, тем выше их вязкость во всем интервале температур. При практически одинаковых строении олигомерной цепи и молекулярной массе, олигодиены с различными функциональными группами значительно отличаются по вязкости, которая зависит от числа функциональной групп и силы межмолекулярного взаимодействия одной группы.
Аналогичные влияния происходят в полимерах на основе олигомеров с концевыми функциональными группами. В результате ИК-спектроскопического исследования установлено, что количество водородносвязанных групп в свежеприготовленных полимерах отличается. Симбатно с количеством водородносвязанных групп в растворе полимеров происходят ассоциативные процессы, что выражается в росте вязкости раствора полимера в неполярных растворителях. После удаления растворителя линейные полимеры не растворимы в обычно применяемых растворителях, в том числе и в тех, в которых получены, происходит физическое гелеобразование (полная потеря текучести).
Методом МУРР показано, что водородные связи являются основной движущей силой ассоциации, следовательно, и сегрегации блоков различной природы сегментированных полимеров.
Олигомеры металлокомплексов
тетра-(2,3-тиофено)порфиразина
, ,
Ивановский государственный химико-технологический университет,
Институт химии растворов РАН, Иваново, Россия, *****@***ru
Методом темплатной полициклотетрамеризации 2,3-дициантиофена и 2,3,4,5-тетрациантиофена в присутствии солей соответствующих металлов и каталитического количества ацетата аммония синтезированы олигомерные металлокомплексы тетра(2,3-тиофено)порфиразина – тиаизолога фталоцианина.
Структура олигомеров определяется соотношением в реакционной смеси ди– и тетрафункционального мономеров.
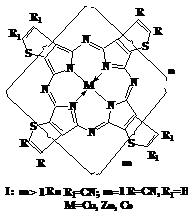 Полученные олигомерные металлокомплексы имеют ряд уникальных особенностей:
Полученные олигомерные металлокомплексы имеют ряд уникальных особенностей:
- жесткая плоскостная структура тетразапорфинового остова элементарного звена;
- возможность введения в состав олигомера различных металов;
- наличие единой π-электронной система всей макромолекулы;
- наличие в составе макролиганда азатиакраун-полостей и подандов нескольких типов, обладающих различной координационной активностью;
- возможность химической модификации олигомера взаимодействием по атомам серы;
Идентификация структуры олигомеров по электронным спектрам поглощения, данным элементного анализа и содержанию концевых групп показала, что полученные соединения представляют собой олигомеры со степенью полимеризации не более 6. Предельные размеры молекул, очевидно, обусловлены термодинамическими причинами и, в частности, потерей ароматичности за счет сильного искажения плоскостной структуры молекулы по мере увеличения ее размеров.
Перспективы использования в качестве катализаторов, комплекситов, сенсоров, полупроводников, материалов для нелинейной оптики и основы супрамолекулярных рецепторов делают полученные олигомеры интересными не только в научном плане.
СИНТЕЗ И СВОЙСТВА РАЗВЕТВЛЕННЫХ АЛКИДУРЕТАНОВЫХ БЛОКСОПОЛИМЕРОВ
Институт химии высокомолекулярных соединений НАН Украины, Киев, Украина, E-mail: *****@***net
Синтезированы разветвленные блоксополимеры на основе алкидных смол и функционализированных изоцианатсодержащих олигомеров различной химической природы, полученных с использованием алифатических, ароматических изоцианатов и растительных масел, соотношение компонентов было различным. Синтез осуществлялся в 2 стадии: первая стадия – приготовление функционализированных изоцианатсодержащих олигомеров, вторая – синтез алкидуретановых блоксополимеров путем химической модификации наполненных и ненаполненных алкидных смол изоцианатными олигомерами.

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 |
