
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Когда этот процесс изображают сокращённо: М ←→ Mn+ + ne–, то может показаться, что это просто ионизация атома. Но есть существенная разница. Энергии ионизации измеряются в газовой фазе, а в данном процессе атомы и ионы не свободные: атомы связаны друг с другом в твёрдую фазу, а ионы – с растворителем. Процесс (1) можно мысленно представить в виде суммы следующих четырёх стадий:
(2) М(тв.) = M(г.); Δ2Н – энергия атомизации
(3) M(г.) = M n+(г) + ne– (г.); Δ3Н = I1+I2+...In – сумма первых n энергий ионизации
(4) M n+(г.) + mH2O(ж.) = [M(OH2)m]n+(ж.); Δ4Н = ΔсольвН – тепловой эффект сольватации;
(5) ne– (г.) = ne– (тв.); Δ5Н = –nW – минус работа выхода электронов из металла (работа выхода W взята с противоположным знаком, т. к. электроны не вырываются из металла, а, наоборот, входят в него).
Тогда для процесса (1) по закону Гесса Δ1Н = Δ2Н + Δ3Н + Δ4Н + Δ5Н, где первые два слагаемых положительны, а два других отрицательны. Следовательно, электродные потенциалы металлов зависят не только от атомных свойств, но и от прочности связи атомов и электронов в простом веществе, и от энергии сольватации, и от работы выхода.
Если электрод инертный (не расходуется и не образуется, как Pt в водородном электроде), то его природа не влияет на равновесный потенциал, но влияет на скорость установления равновесия. Здесь электрод – катализатор. Поэтому часто вместо термина “электродный потенциал” употребляют “окислительно-восстановительный потенциал” или “редокс-потенциал”.
2.2. Концентрации или парциальные давления окисленной и восстановленной форм. Качественно ясно по принципу Ле Шателье: чем выше концентрация окисленной формы и чем ниже концентрация восстановленной, тем сильнее эта система притягивает электроны, т. е. тем выше j. Количественную зависимость дает уравнение В. Нернста (без вывода):
j = jº + (RT/nF) ln([Ox]/[Red]), где
R и T – газовая постоянная и абсолютная температура, n – число электронов, передаваемых в электродной реакции, [Ox] – концентрация (точнее, активность) или давление окисленной формы в степени, соответствующей коэффициенту в уравнении, причем здесь учитывается не только сам окислитель, но вообще все реагенты, стоящие в одной стороне уравнения с окислителем, а [Red] – то же для восстановленной формы, j0 – стандартный электродный потенциал. Как всегда, если вещество составляет отдельную твердую или жидкую фазу, его активность по определению равна единице и не пишется; если вещество растворено, то подставляется его молярная концентрация, а стандартным состоянием является 1 М раствор; если вещество плохо растворимое, то стандартное состояние – его насыщенный раствор, то есть опять-таки равновесие с отдельной фазой растворяемого вещества, а если вещество в газовой фазе, то ставится его парциальное давление в атмосферах, а стандартное состояние – 1 атм. Если все реагенты и продукты – в стандартных состояниях, то под логарифмом единица, второе слагаемое равно нулю, и j = jº. Если подставить коэффициент перехода от натуральных к десятичным логарифмам, числовые значения R и F и принять Т = 298 К, то
j = jº + (0,0591В/n)lg([Ox]/[Red]). Примеры.
j(Zn2+/Zn) = jº + (RT/2F)ln[Zn2+] – без знаменателя;
j(H+/H2) = jº + (RT/2F)ln{[H+]2/p(H2)} или
j(H+/H2) = jº + (RT/F)ln{ [H+]×(p(H2))–1/2}.
Для реакции MnO4– +8H+ +5e ←→ Mn2+ + 4H2O
j = jº + (RT/5F)ln{[MnO4–][H+]8/[Mn2+]}. Заметьте, что число электронов здесь подсчитано по закону сохранения заряда (без них суммарный заряд ионов слева +7, справа +2) и не требует представления о том, что в перманганате степень окисления марганца 7+. Степень окисления – полезное, но условное понятие, а 5е в этой реакции определяются объективно.
Обязательно ли в гальваническом элементе погружать металл в раствор его соли? Вовсе нет, в источниках тока часто используют другие электролиты. Но если мы погружаем цинк в раствор, где концентрация его ионов равна нулю, то получаем из уравнения Нернста потенциал –¥. Смысла в этом нет. Электрод с таким потенциалом мгновенно начинает окисляться, в растворе появляются ионы цинка, их концентрация быстро растет, и потенциал нестабилен. Поэтому для наглядности мы используем заданную и довольно большую концентрацию, которая уже не может сильно измениться.
2.3. Температура. В уравнении Нернста она влияет в двух местах: ведь и стандартный потенциал сам может зависеть от температуры. Однозначно предсказать вид зависимости сложно, но она, безусловно, есть. Поэтому уравнение Нернста лучше записать в виде
j = jºТ + (RT/nF) ln([Ox]/[Red]). Индекс Т подчеркивает, что jº соответствует конкретной температуре. В справочниках чаще всего приводят стандартные потенциалы при 298 К.
2.4. Зависит ли j от способа записи уравнения электродной реакции? Мы знаем, что, если умножить левую и правую части химического уравнения на один и тот же коэффициент b, то соответствующие DН, DS, DG увеличиваются в b раз, а К равновесия возводится в степень b. В уравнении Нернста при этом выражение под логарифмом будет возведено в степень b, а число электронов тоже возрастет в b раз. Если вынести показатель из-под знака логарифма, он сократится – и потенциал останется тем же.
Докажем то же иначе. Убыль энергии Гиббса –DG – это работа химической реакции: –DG = А = nFE. Мы увеличиваем DG и n в одинаковое число раз, E не меняется.
Если поменять местами левую и правую части уравнения, то DН, DS, DG меняют знак, а К равновесия превращается в обратную величину. Но величина j° не изменится. Потенциал характеризует не прямую и не обратную реакцию, а состояние их равновесия. Показания вольтметра не изменятся, если переписать уравнение наоборот. Плюс останется плюсом. Электродный потенциал – это функция состояния, а не процесса. Характеристикой процесса может служить разность электродных потенциалов. При этом, в отличие от внутренней энергии, энтальпии, энтропии, энергии Гиббса, потенциал не зависит и от количества вещества.
Электродные потенциалы не зависят от формы записи уравнения. Но стандартные электродные потенциалы могут зависеть от формы записи, так как там могут подразумеваться разные стандартные состояния. Если в уравнении стоит Cl2(р-р), значит, стандартным состоянием является 1 М раствор, а если Cl2 – то парциальное давление хлора 1 атм. Это разные состояния – разные j. Одно и то же окислительно-восстановительное уравнение можно записать для разных сред, например:
MnO4– + 4H+ +3e ←→ MnO2¯+ 2H2O; j° = 1,69 В;
MnO4– + 2H2O +3e ←→ MnO2¯ + 4 ОН–; j° = 0,60 В.
Почему разные потенциалы? В первом случае под стандартным состоянием понимается состояние с [H+] = 1 моль/л (рН = 0), а во втором – с [ОH–] = 1 моль/л (рОН = 0, а рН = 14). Для любого конкретного рН одно и то же значение j получается из обоих уравнений. Это не разные процессы, а один и тот же, записанный по разным стандартам.
Запишем уравнение Нернста для первой реакции, но подставим туда [H+] = 10–14, а прочие условия оставим стандартными:
j = 1,69 В + 0,0197lg(10–14)4 = 1,69 B – 56*0,0197 = 0,59 В. Отклонение от 0,60 – только от округлений.
2.5. Отклонение от электродного равновесия. До сих пор речь шла о равновесных потенциалах. Но если через электрод идет постоянный ток, то потенциал отклоняется от равновесного. Это явление называется поляризация электрода. Наиболее очевидная (но не единственная) причина поляризации – изменение концентрации реагентов вблизи электрода. У анода возрастает концентрация окисленной формы и уменьшается концентрация восстановленной, и диффузия не успевает их выравнивать по объему раствора, поэтому, согласно уравнению Нернста, потенциал анода увеличивается по сравнению с тем, который соответствует средним по объему концентрациям. И чем больше плотность тока (сила тока на единицу площади), тем сильнее отклонение потенциала от равновесного. На катоде, где идет восстановление, наоборот, растет концентрация восстановленной формы и падает концентрация окисленной, потенциал катода уменьшается. В гальваническом элементе jк>jА, поэтому напряжение U получается меньше эдс. И чем больший ток мы хотим получить от батарейки, тем меньше ее напряжение. Наоборот, при электролизе или при зарядке аккумулятора jк<jА, поэтому, чтобы процесс шел с большой скоростью, приходится прикладывать напряжение больше равновесного (Рис. 1).
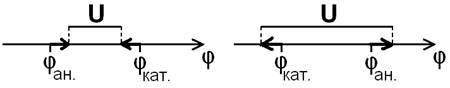
Рис. 1. Схематическое изображение поляризации электродов на шкале потенциалов. φан. и φкат. – равновесные потенциалы анода и катода. Слева – гальванический элемент: реальное напряжение U меньше, чем эдс. Справа – электролиз или зарядка аккумулятора: реальное напряжение U больше равновесного.
2.6. Редокс-потенциалы сложных полуреакций. Если окисление или восстановление идет в несколько стадий, то можно ли суммировать их потенциалы? Закон Гесса применим к изменениям функций состояния, зависящих от количества вещества: DG, DH, DS. Но DG = –nFE = –nF(jox – jred). Поэтому нужно суммировать не потенциалы, а произведения nj для отдельных стадий.
Если 1 и 2 – стадии, а 3 – их сумма, то j1n1 + j2n2 = j3(n1 + n2). Например, равновесие Fe3+ + 3e ←→ Fe неосуществимо на опыте, но соответствующее значение j можно вычислить, зная редокс-потенциалы отдельных стадий:
Fe3+ + e ←→ Fe2+; j°1 = 0,77 В;
Fe2+ + 2e ←→ Fe; j°2 = –0,44 В;
тогда j°3 = (0,77 – 2×0,44)/3 = –0,04 (В).
3. Коррозия
3.1. Общие сведения. Коррозия – разрушение материалов (металлов, пластмасс, керамики...) под влиянием окружающей среды. Мы будем рассматривать только коррозию металлов. Это окислительно-восстановительные процессы. По механизму различают электрохимическую и химическую коррозию. Химическая протекает без участия электролитов при непосредственном контакте металла с окислителем. Типичные случаи – высокотемпературная газовая коррозия, например:
3Fe(тв.) + 2О2(г.) = Fe3O4(тв.) (окалина)

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 |
