

Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Описанные мутантные формы гемоглобина возникают в результате изменений структуры генов по типу замены оснований. Мутации иного характера приводят к появлению аллелей глобинов, обусловливающих другие виды патологии. Так, нарушение процесса рекомбинации между аллельными генами (неравноценный кроссинговер) приводит к изменению числа нуклеотидов в них. Следствием этого может быть сдвиг рамки считывания. Нередким результатом таких структурных изменений генов является подавление синтеза той или иной цепи гемоглобина, приводящее к развитию патологических состояний, известных под общим названием талассемии.
Деления одного нуклеотида в 139-м триплете б-глобинового гена, состоящего из 141 триплета, приводит к сдвигу рамки считывания и прочитыванию в новой рамке терминирующего 142-го кодона. При этом (б-глобиновая цепь удлиняется на пять дополнительных аминокислот. Такой особенностью б-цепей характеризуется гемоглобин Vayne. Когда деления располагается ближе к 5'-концу, активный продукт не синтезируется и развиваются различные формы б-, в - и г-талассемий.
Некоторые варианты гемоглобинов возникают в результате дупли-каций. Так, гемоглобин Grady несет дупликацию 116—118 аминокислотных остатков в г-цепи. В гемоглобине Cranston удлинение р-цепи до 158 аминокислотных остатков является результатом дупликации AG-последовательности после 144-го триплета и последующего сдвига рамки с пропитыванием терминального кодона.
Описанное выше свидетельствует о том, что различные отклонения в структуре ДНК глобиновых генов приводят к замене аминокислот или удлинению полипептидных цепей. Это является причиной образования многих вариантов гемоглобина, которые определяют развитие у человека заболеваний, наследующихся в ряду поколений.
Не меньший интерес представляют механизмы развития различных заболеваний человека, в основе которых лежат мутации генов, приводящие к синтезу белков-ферментов со сниженной активностью или к его подавлению. Это нарушает течение процессов, катализируемых данными ферментами в клетках организма. Примером наследственно детерминированных повреждений метаболизма в организме человека служит фенилкетонурия, развивающаяся вследствие нарушения процессов обмена аминокислоты фенилаланина и накопления в организме токсических промежуточных продуктов.
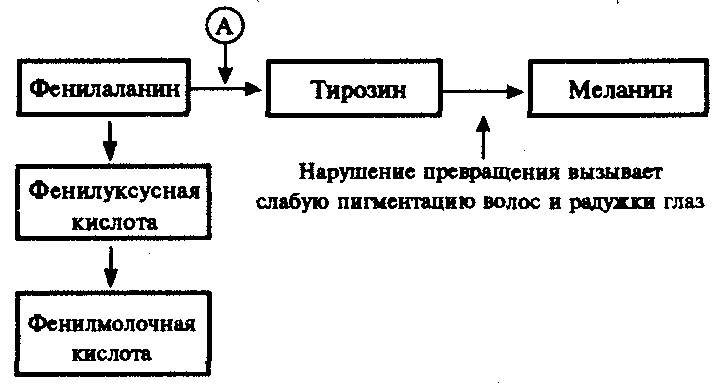
Рис. 4.1. Краткая схема обмена фенилаланина:
А — фермент фенилаланингидроксилаза, наследственный дефект которого приводит к развитию фенилкетонурии
При дефекте фермента фенилаланингидроксилазы фенилаланин не превращается в тирозин (рис. 4.1) и накапливается в крови больных в больших концентрациях (до 0,5—0,6 г/л вместо 0,003— 0,04 г/л в норме). Это приводит к частичному превращению фенилаланина в фенилуксусную и фенилмолочную кислоты, накопление которых наряду с повышенной концентрацией самого фенилаланина оказывает токсическое действие на мозг ребенка. В результате у детей наблюдается различная степень дефекта умственного развития. Нарушение метаболизма фенилаланина сопровождается также нарушением синтеза пигмента меланина, поэтому у больных наблюдается слабая пигментация волос и радужки глаз. Кроме того, высокая концентрация фенилаланина оказывает ингибирующее влияние на ряд ферментных систем, участвующих в превращении других аминокислот: у больных развивается судорожный синдром, нарастает отставание интеллектуального развития. Наследование фенилкетонурии осуществляется по рецессивному типу.
Таким образом, рассмотренные выше примеры демонстрируют весь спектр действия молекулярно-генетических механизмов, обеспечивающих образование в человеческом организме белков как нормально функционирующих, так и обусловливающих развитие различных патологических состояний. Из сказанного по поводу гемоглобина следует, что, во-первых, образование главного функционального белка эритроцитов находится под генным контролем, во-вторых, формирование тетрамерной формы этого белка, с которой связана его физиологическая активность, требует взаимодействия неаллельных генов б - и в-глобинов.
Специфический контроль небелковой части молекулы гемоглобина также имеет место и осуществляется независимо, через гены ферментов, необходимых для синтеза гема. Особенности проявления патологических признаков у носителей мутантных аллелей свидетельствуют о существовании определенных отношений между ними и нормальными аллелями. Так, аллель серповидно-клеточности в сочетании с нормальным аллелем (3-глобина (НbА HbS) проявляет себя в обычных условиях как рецессивный. Так же ведет себя мутантный аллель гена, детерминирующего синтез фермента фенилаланингидроксилазы. Проявлением взаимодействия между мутантным и нормальным аллелями по типу доминирования последнего является формирование в организме белка с нормальными свойствами у гетерозигот. Отсутствие нормального аллеля в генотипе организма, гомозиготного по мутантному аллелю, приводит к развитию патологического состояния, обусловленного нарушением функциональной активности соответствующего белка.
Особую группу наследственно обусловленных патологических состояний у человека представляют заболевания, причиной которых являются мутации митохондриальной ДНК (мгДНК).
Биосинтез митохондриальных белков находится под контролем двух генетических систем: ядерных и митохондриальных генов. Большая часть белков кодируется ядерной ДНК, синтезируется в цитоплазме, а затем транспортируется в митохондрии. Наряду с этим в кольцевой молекуле ДНК органеллы имеются гены, которые отвечают за собственный синтез белков, а также участвующих в нем тРНК и рРНК. В ядерном геноме имеется значительное количество генов, обеспечивающих функционирование митохондриальной ДНК. Предполагают, что мутации некоторых ядерных генов приводят к делениям значительных участков ДНК митохондрии. В результате нарушается синтез собственных белков, к числу которых относятся и ферменты дыхательных цепей, нарушается дыхательная функция митохондрии.
У человека описано более 100 заболеваний, причиной которых являются изменения в структуре мтДНК (см. 6.4.1.4).
4.2. КЛЕТОЧНЫЕ МЕХАНИЗМЫ ОБЕСПЕЧЕНИЯ НАСЛЕДСТВЕННОСТИ И ИЗМЕНЧИВОСТИ
У ЧЕЛОВЕКА
В генетическом материале человека в ряде ситуаций возникают изменения, которые, непосредственно не затрагивая отдельных генов, вызывают серьезные нарушения в состоянии организма. Такие изменения чаще всего касаются структуры хромосом или их числа в клетках. Результатом этого является нарушение баланса генов, т. е. того соотношения доз различных аллелей, которое требуется для нормального развития признаков и организма в целом.
Постоянство кариотипа поддерживается в ряду клеточных поколений благодаря митозу. В ряду поколений организмов это постоянство обеспечивается сочетанием мейоза и оплодотворения. Нарушение митоза и мейоза, обусловливающих закономерное распределение хромосом при образовании соматических и половых клеток, может служить причиной изменения строения и числа этих ядерных структур.
Нередко хромосомные перестройки появляются в результате воздействия на клетки внешних факторов. К таким факторам относится, например, ионизирующее излучение, вызывающее разрывы хромосом и последующие изменения их структуры. У человека описаны также случаи наследственно обусловленной неустойчивости хромосом, их сверхчувствительности к действию агентов различной природы, приводящих к хромосомным разрывам. Это наблюдается при анемии Фанкони, синдроме Блума, атаксии-телеангиэктазии, пигментной ксеродерме. Так, при пигментной ксеродерме высокая чувствительность к ультрафиолетовому свету, сопровождающаяся повышенной ломкостью хромосом, связана с наследственно обусловленным нарушением репарации ДНК.
Изменение числа хромосом, как правило, является результатом нарушения нормального течения клеточных делений, что приводит к образованию анэуплоидных и полиплоидных соматических клеток или гамет с аномальным числом хромосом.
Повреждения механизмов обеспечения наследственности, действующих на клеточном уровне, в масштабе организма приводят к разным результатам. Так, мутации в соматических клетках организма (соматические мутации) могут приводить к различным заболеваниям особи, однако без передачи их потомству при половом размножении. Нарушения наследственной программы в половых клетках (генеративные мутации), не Проявляясь в фенотипе данного организма, ведут к появлению мутантного потомства. Следовательно, точное воспроизведение определенных наследственных характеристик в ряду поколений клеток организма способствует поддержанию здоровья данной особи. Залогом появления здорового в наследственном отношении потомства является в первую очередь сбалансированность генома родительских гамет, содержащего благоприятные аллели генов. При наличии в геноме гаметы одного из родителей «неблагоприятных» аллелей генов их действие может снижаться в результате взаимодействия с нормальными аллелями другого родителя.
4.2.1. Соматические мутации
Мутации различного ранга (генные, хромосомные или геномные), возникающие в соматических клетках организма, наследуются потомками этих клеток и делают организм мозаиком, т. е. особью со смешанными популяциями клеток. В разд. 3.6.5.1 и 3.6.5.2 рассмотрены примеры естественного мозаицизма женского организма по активно функционирующим в его клетках Х-хромосомам и связанное с этим явление аллельного исключения, когда в разных клетках организма экспрессируются разные аллели Х-сцепленных генов.
К примеру, у женщины — гетерозиготной носительницы рецессивного аллеля гемофилии — степень нарушения свертывающей системы крови зависит от соотношения соответствующих клеток с генетически инактивированными Х-хромосомами, несущими нормальный или му-тантный аллель.
Нередко у человека встречается мозаицизм по геномным мутациям, связанный с нарушением расхождения хромосом при митозе. Например, в случае синдрома Дауна (трисомия по 21-й хромосоме) мозаицизм встречается с частотой 2 на 48 пациентов, а в популяции их частота равна 1 на 31 000. Чем раньше в ходе развития организма происходит нарушение деления соматических клеток, сопровождающееся нерасхождением дочерних хромосом к полюсам ахроматинового веретена, тем более выраженной будет симптоматика заболевания, вызываемого данной анэуплоидией. Нарушение митоза на более поздних стадиях индивидуального развития приводит к локальному мозаицизму, который может не сопровождаться выраженными отклонениями от нормы. В этом случае наиболее опасным является мозаицизм клеток генеративных тканей, из которых с достаточно большой вероятностью организм может образовывать гаметы с аномальным числом хромосом.

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 |
