

Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Специфичность проявления хромосомного заболевания определяется изменением содержания определенных структурных генов, кодирующих синтез специфических белков. Так, при болезни Дауна обнаружено повышение в 1,5 раза активности фермента супероксид-дисмутазы I, ген которого располагается в 21-й хромосоме и представлен у больных в трехкратной дозе. Эффект «дозы гена» обнаружен более чем для 30 генов, локализованных в разных хромосомах человека.
Полуспецифические симптомы проявления хромосомных болезней связаны в значительной мере с дисбалансом генов, представленных многими копиями, которые контролируют ключевые процессы в жизнедеятельности клеток и кодируют, к примеру, структуру рРНК, тРНК, гистонов, рибосомальных белков, актина, тубулина.
Неспецифические проявления при хромосомных болезнях связывают с изменением содержания гетерохроматина в клетках, который оказывает влияние на нормальное течение клеточного деления и роста, формирование в онтогенезе количественных признаков, определяемых полигенами.
6.4.1.2. Генные (или менделевские) болезни
К указанным заболеваниям относятся моногенно обусловленные патологические состояния, наследуемые в соответствии с законами Менделя.
В зависимости от функциональной значимости первичных продуктов соответствующих генов генные болезни подразделяют на наследственные нарушения ферментных систем (энзимопатии), дефекты белков крови (гемоглобинопатии), дефекты структурных белков (коллагеновые болезни) и генные болезни с невыясненным первичным биохимическим дефектом.
Энзимопатии. В основе энзимопатии лежат либо изменения активности фермента, либо снижение интенсивности его синтеза. У гетерозигот-носителей мутантного гена присутствие нормального аллеля обеспечивает сохранение около 50% активности фермента по сравнению с нормальным состоянием. Поэтому наследственные дефекты ферментов клинически проявляются у гомозигот, а у гетерозигот недостаточная активность фермента выявляется специальными исследованиями.
В зависимости от характера нарушения обмена веществ в клетках среди энзимопатий различают следующие формы.
1. Наследственные дефекты обмена углеводов (галактоземия — нарушение метаболизма молочного сахара — лактозы; мукополисаха-ридозы — нарушение расщепления полисахаридов).
2. Наследственные дефекты обмена липидов и липопротеинов (сфинголипидозы — нарушение расщепления структурных липидов; нарушения обмена липидов плазмы крови, сопровождающиеся увеличением или снижением в крови холестерина, лецитина).
3. Наследственные дефекты обмена аминокислот (фенилкетонурия —нарушение обмена фенилаланина (см. разд. 4.1); тирозиноз— нарушение обмена тирозина; альбинизм — нарушение синтеза пигмента меланина из тирозина и др.).
4. Наследственные дефекты обмена витаминов (гомоцистинурия — развивается как результат генетического, дефекта кофермента витаминов В6 и B12, наследуется по аутосомно-рецессивному типу).
5. Наследственные дефекты обмена пуриновых и пиримидиновых азотистых оснований (синдром Леша — Найяна, связанный с недостаточностью фермента, который катализирует превращение свободных пуриновых оснований в нуклеотиды, наследуется по Х-сцепленному рецессивному типу).
6. Наследственные дефекты биосинтеза гормонов (адреногенитальный синдром, связанный с мутациями генов, которые контролируют синтез андрогенов; тестикулярная феминизация, при которой не образуются рецепторы андрогенов).
7. Наследственные дефекты ферментов эритроцитов (некоторые гемолитические несфероцитарные анемии, характеризующиеся нормальной структурой гемоглобина, но нарушением ферментной системы, участвующей в анаэробном (бескислородном) расщеплении глюкозы. Наследуются как по аутосомно-рецессивному, так и по Х-сцепленному рецессивному типу).
Гемоглобинопатии. Это группа наследственных заболеваний, вызываемых первичным дефектом пептидных цепей гемоглобина и связанным с этим нарушением его свойств и функций. К ним относят метгемоглобинемии, эритроцитозы, серповидно-клеточную анемию, талассемии (см. § 4.1).
Коллагеновые болезни. В основе возникновения этих заболеваний лежат генетические дефекты биосинтеза и распада коллагена — важнейшего структурного компонента соединительной ткани. К этой группе относят болезнь Эллерса — Данлоса, характеризующуюся большим генетическим полиморфизмом и наследующуюся как по аутосомно-доминантному, так и по аутосомно-рецессивному типу, болезнь Марфана, наследующуюся по аутосомно-доминантному типу, и ряд других заболеваний.
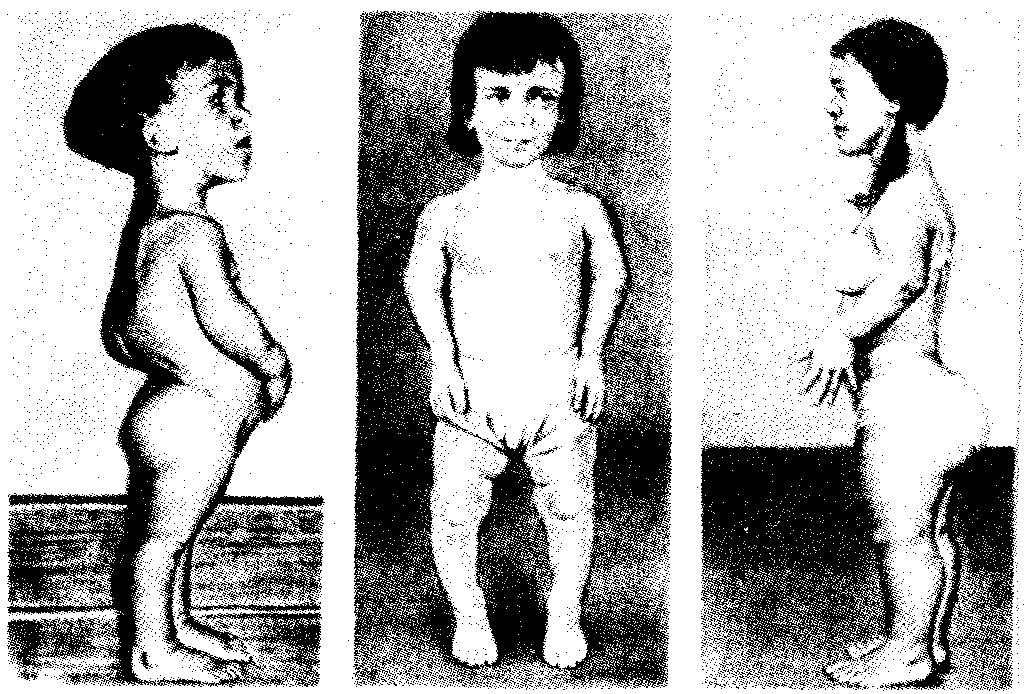
Рис. 6.23. Больные с ахондроплазией
Наследственные болезни с невыясненным первичным биохимическим дефектом. К этой группе принадлежит подавляющее большинство моногенных наследственных болезней. Наиболее распространенными являются следующие.
1. Муковисцидозы — встречаются с частотой 1:2500 новорожденных; наследуются по аутосомно-рецессивному типу. В основе патогенеза заболевания —наследственное поражение экзокринных желез и железистых клеток организма, выделение ими густого, измененного по составу секрета и связанные с этим последствия.
2. Ахондроплазия — заболевание, в 80—95% случаев обусловленное вновь возникшей мутацией; наследуется по аутосомно-доминантному типу; встречается с частотой приблизительно 1:100 000. Это заболевание костной системы, при котором наблюдаются аномалии развития хрящевой ткани преимущественно в эпифизах трубчатых костей и костях основания черепа (рис. 6.23).
3. Мышечные дистрофии (миопатии) —заболевания, связанные с поражением поперечно-полосатых и гладких мышц. Различные формы характеризуются разным типом наследования. Например, прогрессирующая псевдогипертрофическая миопатия Дюшена наследуется по Х-сцепленному рецессивному типу и проявляется преимущественно у мальчиков в начале первого десятилетия жизни. Известна мышечная псевдогипертрофическая дистрофия, наследующаяся по аутосомно-рецессивному типу, которая начинает развиваться во второй половине первого десятилетия жизни и встречается с одинаковой частотой у обоих полов. Мышечная дистрофия плечевого и тазового пояса: наследуется по аутосомно-доминантному типу и т. д.
Генетическое многообразие генных болезней. Изучение наследственных заболеваний у человека свидетельствует о том, что нередко сходное фенотипическое проявление болезни бывает обусловлено несколькими различными мутациями. Это явление впервые было описано в 30-х гг. и названо генетической гетерогенностью наследственных заболеваний. Генетическая гетерогенность наследственных болезней может быть обусловлена мутациями разных генов, кодирующих ферменты одного метаболического пути, а также мутациями одного и того же гена, приводящими к появлению разных его аллелей.
Среди рассмотренных выше наследственных болезней особенно высокой степенью генетического полиморфизма отличаются мукопо-лисахаридозы, генетическая разнородность которых объясняется множественными мутациями в 11—12 генах, связанных общей функцией расщепления полисахаридов. Большой генетической гетерогенностью характеризуется врожденная аутосомно-рецессивная форма глухоты, при которой различают не менее 35 генетически различных вариантов с фенотипически сходным проявлением.
Большие перспективы в расшифровке наследственной гетерогенности генных болезней открываются в связи с применением молекулярно-генетических методов их прямого анализа с помощью ДНК-зондов.
Клиническое многообразие наследственных болезней. Разнообразие клиники наследственных болезней проявляется в различии времени начала заболевания, в спектре и степени выраженности симптомов, в течении и исходе у разных больных. Например, наследуемая по аутосомно-доминантному типу хорея Гентингтона, при которой поражаются базальные ганглии головного мозга, клинически начинает проявляться в виде непроизвольных движений в разном возрасте, но чаще в 40—45 лет. С временем начала клинического проявления связана и тяжесть течения заболевания (см. 6.4.1.4).
О клиническом полиморфизме можно говорить лишь в отношении генетически определенной наследственной формы. Причины клинического полиморфизма могут быть как генетическими, так и средовы-ми. К генетическим причинам можно отнести действие генов-модификаторов на проявление патологически измененного гена и сложную систему разнообразных взаимодействий между ним и другими генами. Кроме того, разнообразие клинического проявления наследственных заболеваний может зависеть от факторов среды, в которой развивается организм и которая влияет на проявление патологически измененных генов.
6.4.1.3. Мультифакториальные заболевания,
или болезни с наследственным предрасположением
Эта группа болезней отличается от генных болезней тем, что для своего проявления нуждается в действии факторов внешней среды. Среди них также различают моногенные, при которых наследственная предрасположенность обусловлена одним патологически измененным геном, и полигенные. Последние определяются многими генами, которые в нормальном состоянии, но при определенном взаимодействии между собой и с факторами среды создают предрасположение к появлению заболевания. Они называются мультифакториальными заболеваниями (МФЗ).
Заболевания моногенные с наследственным предрасположением относительно немногочисленны. К ним применим метод менделевского генетического анализа. Учитывая важную роль среды в их проявлении, они рассматриваются как наследственно обусловленные патологические реакции на действие различных внешних факторов (лекарственных препаратов, пищевых добавок, физических и биологических агентов), в основе которых лежит наследственная недостаточность некоторых ферментов.
К таким реакциям могут быть отнесены наследственно обусловленная непереносимость сульфаниламидных препаратов, проявляющаяся в гемолизе эритроцитов, повышении температуры при применении общих анестезирующих средств.
У человека описана мутация, обусловливающая патологическую реакцию на загрязнение атмосферы, которая проявляется в раннем развитии эмфиземы легких (в возрасте 30—40 лет). У генетически чувствительных индивидов нежелательные реакции могут вызывать некоторые компоненты пищи и пищевые добавки. Известна непереносимость у ряда людей молочного сахара —лактозы. Гены непереносимости лактозы широко распространены среди азиатского населения (до 95—100%) и среди американских негров и индейцев (до 70—75%). У некоторых людей наблюдается непереносимость к употребляемым в пищу конским бобам, вызывающим у них гемолиз. Ряд лиц не переносит жирной пищи и в раннем возрасте страдает атеросклерозом, что повышает риск инфаркта миокарда. У некоторых людей употребление в пищу сыра и шоколада провоцирует мигрень. Отмечены специфические реакции людей на алкоголь. Консерванты и пищевые красители у некоторых людей не подвергаются нормальному усвоению, что также проявляется в непереносимости этих компонентов пищи.

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 |
