

Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
in_coefs = new Texture2D(gpu, 2, 2, 1, TextureUsage. None, SurfaceFormat. Vector4);
float[] data = new float[2 * 2 * 4];
for (int u = 0; u < 2; u++)
for (int v = 0; v < 2; v++) //здесь 2 цикла могли быть заменены одним по i от 0
//до <количества пар> - 1
{
int i = (u * 2 + v) * 4;
data[i] = (float)coefs[i++];
data[i] = (float)coefs[i++];
data[i] = (float)coefs[i++];
data[i] = (float)coefs[i++];
}
in_coefs. SetData<float>(data); //in_coefs - переменная типа Texture2D
//Задаем квад (вершинные и текстурные координаты, соответственно).
//Y-координата текстуры инвертирована по отношению к Y-координате вершины,
//т. к. ось Y направлена снизу вверх в пространстве вершин и сверху вниз в пространстве экрана.
float du = 0.5f / w, dv = 0.5f / h;
// смещения для адресации текселей по центрам
//(в случае текстур с размерами не равными степени числа 2
quad = new VertexBuffer(gpu, typeof(VertexPositionTexture), 4, BufferUsage. None); //переменная типа VertexBuffer
quad. SetData<VertexPositionTexture>(new VertexPositionTexture[] {
new VertexPositionTexture(new Vector3(-1, 1, 0), new Vector2(0 + du, 0 + dv)),
new VertexPositionTexture(new Vector3( 1, 1, 0), new Vector2(1 + du, 0 + dv)),
new VertexPositionTexture(new Vector3(-1, -1, 0), new Vector2(0 + du, 1 + dv)),
new VertexPositionTexture(new Vector3( 1, -1, 0), new Vector2(1 + du, 1 + dv))
}); // кубу в пространстве сопоставлены смещенные углы плоскости
gpu. Vertices[0].SetSource(quad, 0, VertexPositionTexture. SizeInBytes); //размер квада
gpu. VertexDeclaration = new VertexDeclaration(gpu, VertexPositionTexture. VertexElements); //неясно, что
//Задаем шейдер и его входные параметры.
//Компиляция текстового файла с шейдером
CompiledEffect e = pileEffectFromFile("force. fx", null, null, CompilerOptions. None, TargetPlatform. Windows);
//Проверка успешности компиляции
if (!e. Success) throw new Exception(e. ErrorsAndWarnings);
//Записывает программу эффекта в память видеокарты
fx = new Effect(gpu, e. GetEffectCode(), CompilerOptions. None, null); //переменная типа Effect
//Выбирает роцедуру из кода эффекта, которую надо будет выполнять
fx. CurrentTechnique = fx. Techniques["Force"];
//Передача на видеокарту координат частиц
fx. Parameters["in_pos"].SetValue(in_pos); //видимо, in_pos и in_coefs описаны в шейдере как входные текстуры
//Передача на видеокарту параметров потенциалов взаимодействия
fx. Parameters["in_coefs"].SetValue(in_coefs);
}
private void ForceGPU()
{
int i, u, v;
//Задаем текстуру с координатами и типами частиц
float[] data = new float[w * h * 4];
/* Текстура представляет собой таблицу с размером w по горизонтали и h по вертикали.
* В каждой ячейке таблицы находится 4-х-вектор, хранящий координаты и тип ионов.
* Номер частицы в линейном массиве i вычисляется по формуле i = v * w + u
*/
for (v = 0; v < h; v++)
for (u = 0; u < w; u++)
{
i = v * w + u;
data[i * 4 + 0] = (float)pos[i * 3 + 0];
data[i * 4 + 1] = (float)pos[i * 3 + 1];
data[i * 4 + 2] = (float)pos[i * 3 + 2];
data[i * 4 + 3] = type[i] / 2f;
}
in_pos. SetData<float>(data); //in_pos - переменная типа Texture2D
//Задаем новую рендер-цель, сохраняя старую.
RenderTarget2D old_render_target = (RenderTarget2D)gpu. GetRenderTarget(0); //Зачем нужно преобразование типа?
gpu. SetRenderTarget(0, out_force); //out_force - переменная типа RenderTarget2D
gpu. Clear(new Color(0, 0, 0, 0)); //Обнуление сил в рендер-цели
fx. Begin(); //спросить, что делает Begin. Загружает в память?
for (i = 0; i < ions; i++)
{
//Обновляем ссылку на рендер-цель (необходимо для корректного выполнения операции += в шейдере)
gpu. SetRenderTarget(0, null); fx. Parameters["out_force"].SetValue(out_force. GetTexture());
gpu. SetRenderTarget(0, out_force);
//Передаем в шейдер текстурные координаты i-й частицы.
u = i % w; v = i / w; //очевидно, % - остаток от деления
fx. Parameters["u_i"].SetValue((u + 0.5f) / w); //передаётся центр ячейки
fx. Parameters["v_i"].SetValue((v + 0.5f) / h); //передаётся центр ячейки
//Вычисляем взаимодействие i-й частицы со всеми остальными.
fx. CurrentTechnique. Passes["force_i"].Begin();
gpu. DrawPrimitives(PrimitiveType. TriangleStrip, 0, 2);
fx. CurrentTechnique. Passes["force_i"].End();
}
fx. End(); //В цикле по всем частицам заполняется одна и та же рендер-цель
//Восстанавливаем старую рендер-цель.
gpu. SetRenderTarget(0, old_render_target);
//Получаем результат из новой рендер-цели
data = new float[w * h * 4];
for (i = 0; i < 3; i++) gpu. Textures[i] = null;
out_force. GetTexture().GetData<float>(data); //out_force - массив с рендер-целью
for (v = 0; v < h; v++)
for (u = 0; u < w; u++)
{
i = v * w + u;
force[i * 3 + 0] = data[i * 4 + 0];
force[i * 3 + 1] = data[i * 4 + 1];
force[i * 3 + 2] = data[i * 4 + 2];
}
}
7.4. Постановка граничных условий и стабилизация макросостояния молекулярно-динамической системы
В настоящем пособии мы рассматриваем только моделирование кристаллов с нулевыми граничными условиями, т. е. – кристаллитов с конечным числом частиц в вакууме. Применение таких граничных условий в идеале не требует для расчёта сил, скоростей и перемещений ионов никаких других уравнений, кроме соотношений -. Однако, накопление вычислительных погрешностей приводит к нарушениям законов сохранения импульса, момента импульса и энергии, что может приводить к появлению движения и вращения кристалла как целого, а также к росту его температуры. Для исключения этих эффектов можно использовать следующие методы.
7.4.1. Компенсация импульса и момента импульса
Импульс модельного кристалла из N частиц даётся формулой
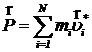 ,
,
где mi – массы частиц, ![]() - их скорости до компенсации импульса. Скорость центра инерции кристалла связана с импульсом соотношением
- их скорости до компенсации импульса. Скорость центра инерции кристалла связана с импульсом соотношением
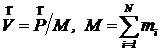 .
.
Вычитание этой скорости из скоростей всех частиц останавливает собственное движение кристалла:
![]() .
.
Предположим, что кристалл только вращается как твердое тело вокруг какой-нибудь оси, и больше частицы никуда не движутся (для упрощения выражений). Это вращение характеризуется постоянной угловой скоростью ![]() , связанной со скоростями частиц соотношением
, связанной со скоростями частиц соотношением
![]() .
.
Здесь ![]() - скорости и координаты i-ой частицы. Момент импульса этого кристалла рассчитывается по формуле
- скорости и координаты i-ой частицы. Момент импульса этого кристалла рассчитывается по формуле
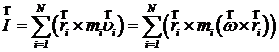 ,
,
где N – количество частиц. Двойное векторное произведение можно преобразовать по формуле
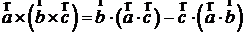 ,
,
тогда получится, что
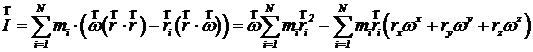 .
.
Это векторное уравнение эквивалентно системе из трех уравнений для компонент векторов ![]() и
и ![]() :
:
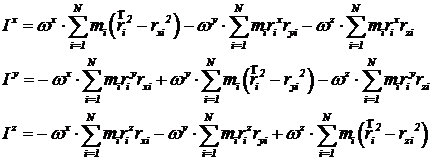 .
.
Значения компонент векторов с верхними и нижними индексами одинаковы – положение индексов указывает на ко - и контравариантность векторов. Отметим также, что слагаемые вида
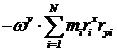
для симметричного относительно оси вращения кристалла равны нулю, так что и для реальных кристаллов должны быть близки к нулю.
Таким образом, если для кристалла с произвольными скоростями частиц рассчитан момент импульса ![]() , то может быть, решением записанной системы уравнений, получена соответствующая угловая скорость
, то может быть, решением записанной системы уравнений, получена соответствующая угловая скорость ![]() . Если теперь получить новые скорости частиц
. Если теперь получить новые скорости частиц ![]() по формуле
по формуле
![]() ,
,
то момент импульса кристалла станет равным нулю. При этом изменение самого импульса будет иметь вид
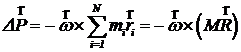 ,
,
где M – масса кристалла, ![]() - радиус-вектор центра инерции. Очевидно, что если отсчитывать координаты
- радиус-вектор центра инерции. Очевидно, что если отсчитывать координаты ![]() от центра инерции, то
от центра инерции, то ![]() , и импульс не изменится. Кроме того, перед остановкой вращения кристалла нужно обнулить суммарный импульс, тогда ось вращения обязательно будет проходить через центр инерции, что подразумевается в формуле.
, и импульс не изменится. Кроме того, перед остановкой вращения кристалла нужно обнулить суммарный импульс, тогда ось вращения обязательно будет проходить через центр инерции, что подразумевается в формуле.
7.4.2. Стабилизация температуры
Стабилизация температуры моделируемой системы в принципе может быть необходима по двум причинам:
· образование дефектов кристаллической решётки, суперионный переход и плавление требуют энергии, так что в изолированной системе происходит повышение потенциальной энергии за счёт понижения температуры; поэтому, для моделирования кристаллов с постоянной температурой необходим алгоритм стабилизации температуры, моделирующий взаимодействие с термостатом;
· опыт показывает, что накопление вычислительной погрешности приводит к медленному, но существенному при больших временах моделирования разогреву кристаллитов; для предотвращения разогрева тоже необходима стабилизация температуры.
Идея стабилизации состоит в том, что в уравнения движения изолированной системы добавляется дополнительное слагаемое, понижающее скорости частиц в случае, если температура системы превосходит заданную и увеличивающее скорости в противоположном случае.
Модификация скоростей производится умножением импульсов на коэффициент px/Q, одинаковый для всех частиц, так что она не приводит к возникновению движения или вращения системы как целого.
Применение стабилизирующей температуру поправки в форме - было предложено Рябовым. Из его работ следует, что в форме - уравнения движения системы соответствуют гамильтониану, совпадающему с выражением для энтальпии H, так что они физически корректно описывают кристаллиты при нулевом (пренебрежимо малом) внешнем давлении и постоянной температуре.
Таким образом, для термостатирования системы с нулевыми граничными условиями можно использовать следующие уравнения движения молекул:
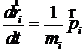 ;
;
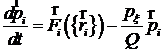 ;
;
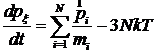 .
.
Здесь N – количество частиц в системе; T – требуемая температура; px - дополнительная переменная размерности Дж×с, связанная с термостатом, Q – величина размерности Дж×с2, задающая частоту колебаний температуры; индекс x не имеет самостоятельного значения, он только показывает, что px - переменная, относящаяся к стабилизации температуры.
В начале моделирования можно принять, что px = 0. На последующих шагах моделирования эта величина изменяется по формуле
![]() .
.
Значение константы Q в принципе произвольно, однако этим значением определяются период и амплитуда колебаний задаваемой температуры. Оценить необходимое значение Q можно из следующих соображений.
Из-за слагаемого, соответствующего термостату, к скоростям частиц добавляется поправка
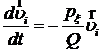 .
.
Если домножить обе части уравнения на ![]() и просуммировать по всем i, то получится, что
и просуммировать по всем i, то получится, что
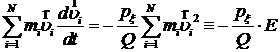 ,
,
где E – удвоенная кинетическая энергия системы. Её требуемое значение как раз и равно 3NkT. Учтём то, что
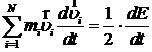
и то, что, в соответствии с формулами и,
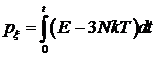 .
.
Таким образом, временная зависимость отклонения величины E от 3NkT даётся уравнением
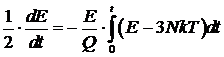 .
.
Можно переписать уравнение в приближённой форме, считая, что E » 3NkT; это верно, если температура колеблется не сильно. Кроме того, вводим новую переменную
![]() ,
,
причём
![]() .
.
Теперь, дифференцируя обе части уравнения по времени, в приближении E » 3NkT получаем
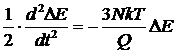 .
.
Это – уравнение гармонических колебаний с циклической частотой
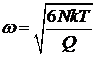 ,
,
откуда
![]() ,
,
где t - период колебаний.
Выражение позволяет выбирать значение Q, исходя из требований, предъявляемых к периоду колебаний. Например так, чтобы период колебаний был существенно меньшим, чем время моделирования.
8. Высокоскоростное моделирование систем с дальнодействием
8.1. Актуальность моделирования
Для получения статистических характеристик кинетических процессов в вычислительных экспериментах необходимо длительное моделирование на системах с большим числом частиц. Однако, как известно, вычислительная нагрузка при моделировании систем с дальнодействующими силами, в которых необходимо учитывать взаимодействие всех частиц между собой, увеличивается квадратично с ростом их числа. Следовательно, представляет интерес разработка высокоскоростных методов, которые позволяли бы проводить длительные вычислительные эксперименты на системах, достаточно больших для получения искомых статистических макропараметров, в реальном времени.
В качестве объекта исследования мы рассмотрим ионный кристалл диоксида урана, моделирование которого актуально в связи с потребностью в прогнозировании свойств окисного ядерного топлива современных реакторов. Для такого прогнозирования необходима, в частности, информация о процессах массопереноса собственных ионов UO2 в широком диапазоне температур, так как эти процессы определяют кинетику изменения состава и свойств. Однако, экспериментальные данные из-за технической сложности измерений в рабочей зоне реактора (высокие температуры, давления, радиоактивность) известны для достаточно низких температур, соответствующих кристаллическому состоянию UO2, что делает невозможным их использование в области суперионной фазы и тем более в расплавленном состоянии. С другой стороны, известные эксперименты по компьютерному моделированию UO2 проводились на небольших системах, при малом числе шагов и существенных затратах времени, что препятствовало получению достоверной информации о процессах массопереноса при температурах ниже точки плавления, в особенности для малоподвижных ионов урана.
Данный раздел посвящен анализу возможностей высокоскоростных алгоритмов моделирования систем с дальнодействием и моделированию кинетических процессов в диоксиде урана высокоскоростной реализацией метода молекулярной динамики (ММД) на системах порядка ~104 частиц, интервалах времени порядка ~107 шагов, в широком диапазоне температур, охватывающем кристаллическое состояние, суперионную фазу и расплав.
8.2. высокоскоростные алгоритмы моделирования систем с дальнодействующими силами
Рассмотрим систему N классических частиц с массами mi и зарядами qi, динамика которой подчиняется уравнениям движения Ньютона, а взаимодействие зарядов системы с единичным зарядом определяется Кулоновским (дальнодействующим) потенциалом U:
mid2ri/dt2 = –qi∂U/∂rij
U = ∑j>i qj|ri – rj|–1
Простейшим методом ее моделирования является прямой расчет сил суммированием парных взаимодействий всех частиц системы друг с другом, с последующим численным интегрированием уравнений движения. Так как в такой системе каждая частица взаимодействует со всеми остальными, то число операций пропорционально N×(N–1). С помощью 3-го закона Ньютона (действие равно противодействию), можно учесть симметричность вклада каждого парного взаимодействия в суммарные силы, действующие на частицы, и сократить число операций вдвое, однократно рассчитывая этот вклад для каждой пары. Но такая оптимизация все же не устраняет квадратичную зависимость вычислительной нагрузки от количества частиц в системе, т. е. увеличение количества частиц в моделируемой системе в 10 раз все равно приводит к 100-кратному увеличению времени расчетов.
Активные попытки преодоления этого вычислительного барьера привели к появлению большого числа численных методов, в которых с целью экономии числа операций используется замена точного прямого расчета сил различными приближениями, а также всевозможные алгоритмические усовершенствования (см. обзорные работы [17], [18]). Условно их можно подразделить на 3 категории: суммирование Эвальда, сеточные методы и мультипольные или иерархические.
Метод Эвальда [19] позволяет рассчитать взаимодействие частиц некоторой «ячейки» (ограниченной области), с ее отражениями, бесконечно транслированными по одному или нескольким измерениям пространства, разделив медленно сходящийся ряд Кулона такой системы:
U = ∑n∑j qj|rij+nL|–1
на сумму двух быстро сходящихся рядов в реальном и «обратном» пространствах:
U = ∑n∑j qj|rij+nL|–1erfc(α|rij+nL|) +
+ 4πL–3∑k≠0∑j qj exp[–ikrij – k2(2α)–2] – qi(2α)/π1/2
Здесь L – период решетки, α – произвольный, но существенный параметр, отвечающий за сходимость обоих рядов суммы, а последнее слагаемое – «потенциал самодействия», нивелирующий соответствующий вклад от ряда в «обратном» пространстве. Область применение метода строго ограничена периодическими системами, однако он широко используется в исследованиях, где важно исключить поверхностные эффекты, неизбежно возникающие при моделировании небольших изолированных систем. В работе Перрама и др. [20] показано, что существует оптимальный выбор параметров, при котором вычислительная нагрузка метода Эвальда растет пропорционально N3/2.
Сеточные методы раскладывают заряд частиц по узлам дискретной сетки, затем решают на ней уравнение поля, используя различные быстрые алгоритмы (быстрое преобразование Фурье, метод Гаусса-Зейделя, последовательной релаксации), а потом интерполируют силы, рассчитанные в узлах сетки, чтобы получить силы, действующие на отдельные частицы. Основная вычислительная нагрузка связана с решением уравнений поля и для большинства алгоритмов пропорциональна NGlgNG, где NG – количество узлов сетки. Областью эффективного применения сеточных методов так же являются периодические системы, так как для моделирования изолированной системы ее приходится окружать зеркальными копиями, чтобы скомпенсировать вклад транслированных отражений. Подробный анализ многочисленных методов этой категории выходит за рамки данной работы (см., например, две монографии по теме [21] и [22]).
Иерархические методы предназначены для быстрого моделирования открытых (изолированных) систем и применяются везде, где невозможно использование периодических граничных условий, например, в динамике галактик, электронных пучков, больших протеинов или любых других систем со сложной геометрией. Они сравнительно немногочисленны, однако, революционизировали область расчетов систем, где «все взаимодействуют со всеми» в значительно большей степени, чем упомянутые выше специализированные «периодические» методы.
Вычислительная «неэффективность» прямого расчета сил связана, в частности, с отсутствием учета «пространственной когерентности» парных взаимодействий данной частицы с локализованными «дальними партнерами» – каждое парное взаимодействие затрачивает одинаковый вычислительный ресурс, независимо от малости вклада в суммарную силу, который убывает обратно пропорционально квадрату расстояния. Однако, прямое отбрасывание дальнодействия (например, введением некоторого радиуса обрезания) оправдано только в ряде частных случаев, где потенциал условно можно считать короткодействующим; в случае Кулоновского взаимодействия ошибки накапливаются относительно быстро.
В 1986 году Барнс и Хат опубликовали алгоритм TreeСode [23], в котором физическое пространство иерархически подразделялось на области для учета относительного положения частиц. Результирующая древесная структура (рис. 7.1), затем использовалась для группировки удаленных частиц в «кластеры», с заменой всех взаимодействий частица–частица одним взаимодействием частица–кластер, существенно уменьшая число операций.
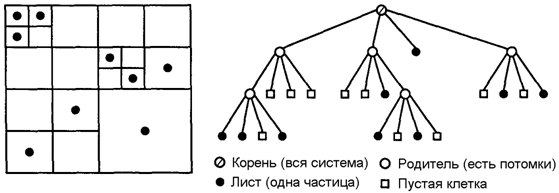
Рис. 7.1. Древесная структура quadtree (двумерный случай).
Основой всех иерархических методов является древовидное представление физического пространства. Корень дерева представляет клетку, которая содержит все частицы системы. Дерево строится рекурсивно – в двухмерном варианте сначала делят корневую клетку на четыре клетки-потомка, а затем дробят полученные клетки, если количество частиц в них больше некоторого фиксированного числа. Полученное таким образом дерево называется деревом квадрантов (quadtree), поскольку у каждой клетки, не являющейся «листовой», по четыре клетки-потомка. Форма дерева соответствует распределению частиц в пространстве, поскольку дерево имеет больше уровней в тех областях, где больше частиц. В трехмерном пространстве родительские клетки делятся на восемь потомков, и структура данных называется деревом октантов (octree).
Как только дерево построено, его узлы можно наполнить информацией об их физическом содержании. Для этого проходом «вверх» (от листьев к корню) ищется центр зарядов каждой клетки и несколько первых членов разложения по мультипольным моментам относительно него:
rc = åri|qi| / å|qi|
M = åqi
Dx = åqirix
Qxy = åqi(3rixriy − ri2δxy)
Здесь rc – центр зарядов клетки, rix – х-компонента расстояния между i-той частицей и центром зарядов, M – монопольный момент распределения зарядов клетки, Dx и Qxy – соответствующие компоненты дипольного и квадрупольного моментов, δxy – символ Кронекера.
Затем проходом «вниз» (от корня к листьям) составляются «списки взаимодействий», когда для каждой частицы данной клетки определяется допустимость расчета ее взаимодействия с частицами «дальней» клетки как с кластером, на основе некоторого «критерия допустимости». Простейшим критерием является представленное в оригинальной работе отношение S/D – длины ребра клетки-кластера S к расстоянию до взаимодействующей частицы D (рис. 7.2).
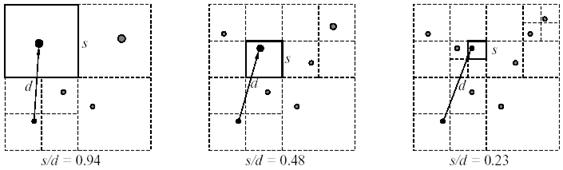
Рис. 7.2. Отношение S/D для разных уровней дерева.
Если отношение меньше некоторого заданного параметра θ, регулирующего точность метода, то мультипольным разложением этой «дальней» клетки-кластера аппроксимируется действие всех вложенных в нее клеток-потомков на рассматриваемую частицу (R – расстояние от частицы до центра зарядов «дальней» клетки):
U = MR–1 + R–3årxDx + R–5åårxryQxy / 2
иначе по тому же критерию проверяются клетки-потомки. Взаимодействие с частицами клеток данной ветви дерева, которые не удовлетворяют критерию – считается прямым способом. Если задать параметр точности θ равным нулю, все частицы системы будут считаться прямым способом, на практике θ выбирают в диапазоне 0.3 – 1.0, в зависимости от задачи.
На каждом временном шаге дерево перестраивается, поскольку частицы перемещаются, и, следовательно, их распределение меняется. Однако, перемещения относительно малы, поэтому дерево можно перестраивать, когда частицы перемещаются за пределы своей клетки. В любом случае центры зарядов и мультипольные разложения всех клеток нужно пересчитывать на каждом временном шаге. Число узлов в дереве октантов равно log2N1/3 = lgN/3lg2 ~ lgN, поэтому построение дерева требует порядка NlgN операций. Также было показано [24], что количество взаимодействий выделенной частицы с однородным сферическим облаком пропорционально lgN/θ2, таким образом, вычисление сил в системе из N частиц также требует порядка NlgN операций.
Наличие дополнительных операции в алгоритме TreeСode, отсутствующих в прямом расчете: построение и заполнение дерева и списков взаимодействий частиц, расчет центров зарядов и мультипольных разложений, однозначно говорит о том, что метод будет эффективнее прямого расчета лишь при количествах частиц в системе, превышающих некоторое значение, которое определяется в основном выбором параметра точности θ.
Также следует упомянуть, что алгоритм TreeСode не учитывает симметрию сил между частицами, т. е. в фазе вычисления сил каждая частица рассматривается отдельно (т. к. если пытаться отслеживать все уже вычисленные пары сил, такой учет получается слишком громоздким). Помимо лишней вычислительной нагрузки это приводит к тому, что нарушается закон сохранения общего импульса и момента импульса системы.
В 1987 году, вскоре после появления TreeСode, Грингард и Рохлин опубликовали альтернативный быстрый мультипольный метод – Fast Multipole Method (FMM) [25], который может рассматриваться как элегантное усовершенствование алгоритма TreeСode, однако, был разработан независимо. В настоящее время FMM входит в десятку лучших алгоритмов 20-го века и широко используется во многих областях физики, математики и компьютерных наук. Алгоритм FMM основан на том факте, что мультипольное разложение с бесконечным порядком членов содержит полную информацию о распределении зарядов в пространстве. Как и в методе TreeСode взаимодействие между ближайшими соседями вычисляется прямым расчетом, а дальние частицы рассматриваются группами, однако, эти вклады считаются по-другому.
Первое основное отличие FMM – дальнодействие рассматривается как единый общий вклад «дальнего поля», который вычисляется как мультипольное разложение высокого порядка. Метод TreeСode рассматривает только взаимодействия типа частица-частица и частица-клетка, в FMM помимо этого вычисляются взаимодействия типа клетка-клетка, как было показано в оригинальной работе [25], это позволяет получить алгоритм с линейной зависимостью вычислительной нагрузки от числа частиц.
FMM a priori позволяет получить произвольную точность (в пределах машинной), если используется мультипольного разложение достаточного порядка, поэтому существенным является выбор наименее ресурсоемкого численного представления мультипольных разложений, операторов их комбинации и трансляции их центров. Первые варианты FMM были двумерны, ориентированы на однородное распределение зарядов и использовали сложную мат. модель представления полей и потенциалов [25], позднее была сформулирована адаптивная трехмерная реализация, использующая сферические гармоники [26].
Второе основное отличие FMM от TreeСode состоит в способе, по которому управляют точностью аппроксимации сил. Метод TreeСode представляет систему в виде дерева мультипольных разложений в центрах зарядов отдельных клеток, и определяет допустимость аппроксимации отношением расстояния между рассматриваемой частицей и центром заряда удаленной клетки к длине ее ребра. В FMM используется иерархия вложенных сеток, а распределение заряда в клетке представляется с помощью мультипольного разложения около геометрического центра клетки, а также используется другой критерий допустимости мультипольного разложения: считается, что две клетки достаточно удалены друг от друга, если расстояние между ними превосходит длину ребра клетки на данном уровне иерархии. Таким образом, аппроксимации в FMM используются намного чаще, чем в методе TreeСode, но это компенсируется более точным описанием распределения зарядов внутри клетки.
Кратко алгоритм FMM устроен следующим образом (см. рис. 7.3):
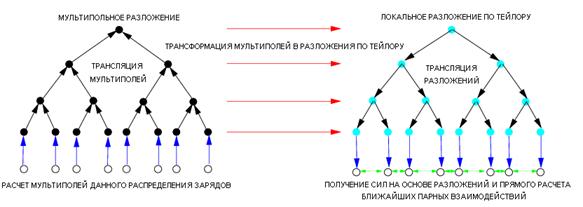
Рис. 7.3. Упрощенная схема алгоритма FMM.
Построение сеточной иерархии, делением клеток на потомки, начиная с корневой, включающей все частицы системы – проход «вниз» (от корня к листьям).
Расчет мультипольных разложений для клеток нижнего уровня и последовательная комбинация мультиполей клеток-потомков со сдвигом центров разложений для получения мультиполей клеток более высоких уровней – проход «вверх» (от листьев к корню). В результате этой фазы в корневой клетке находится суммарное мультипольное разложение всей системы, которое алгоритм TreeСode использует для вычисления сил действующих на частицы.
Трансформация мультипольных разложений всех клеток в локальные (разложения в ряд Тейлора около центров клеток), применение локального разложения каждой клетки к клеткам из ее списка взаимодействий. Трансляция (с добавлением) локальных разложений клеток с верхних уровней своим клеткам-потомкам – проход «вниз» (от корня к листьям). Эта дополнительная фаза реализует взаимодействие клетка-клетка, в результате в каждой клетке самого нижнего уровня будет находиться локальное разложение поля всей системы зарядов около геометрического центра данной клетки.
Вычисление сил, действующих на частицы данной клетки, на основе локального разложения в ряд Тейлора поля системы зарядов около ее геометрического центра и прямого парного взаимодействия с частицами внутри данной клетки и ее ближайших клеток-соседей (для которых мультипольное разложение считалось неприменимым).
На первый взгляд, FMM, с его теоретически предсказываемой линейной зависимостью времени расчетов от числа частиц, превосходит все остальные методы. Однако, этот алгоритм содержит существенную дополнительную вычислительную нагрузку, связанную с расчетом мультипольных и локальных разложений и их трансформацией и трансляциями, поэтому на практике вопрос ставится иначе: при каком числе частиц стоит использовать FMM вместо TreeCode или прямого расчета? Ответ на него не прост, так как время расчетов зависит не только от числа частиц, но и от требуемой точности вычислений.
В алгоритме TreeСode точность определяется параметром θ, для FMM ту же роль играют число членов L в разложениях и глубина R иерархической структуры, Шмидт и Ли показали [27], что его полиномиальная зависимость от N, L и R такова:
P = aBL2 + bNL2 + cBL4 + dB∙(g1 + g2(N/B) + g3(N/B)2).
Здесь a, b, c, g1, g2, g3 – машинно-зависимые параметры, а B = 8R количество клеток на нижнем уровне иерархии R. Это выражение показывает, что отношение (N/B) должно быть мало, чтобы квадратичной зависимостью можно было пренебречь в пользу линейной – на практике это означает увеличение глубины сеточной иерархии с увеличением числа частиц.
Отметим также, что одно из предположений оригинального варианта FMM – более-менее однородное распределение зарядов в пространстве; существенная неоднородность потребует одновременно большей глубины иерархии и большего числа членов в разложениях, что приведет к увеличению вычислительной нагрузки. Поэтому FMM в оригинальной форме плохо применим к неоднородным и динамичным системам, где возможны резкие изменения плотности с течением времени. Для решения этого вопроса были разработаны адаптивные методы [26], в большинстве своем являющиеся гибридами TreeCode и FMM.
Приведем график (см. рис. 7.4) сравнения производительности разных алгоритмов: TreeCode, FMM и прямого расчета из работы [17], данные производительности в этой работе были получены на вычислительной системе SunBlade Sparc, на модели с однородным начальным распределением зарядов, там же приводим данные производительности нашей собственной поточно-параллельной версии прямого расчета. Для метода TreeСode, практически независимо от числа частиц в системе, выбор параметра θ равным 0.2 и 0.5 дает порядок ошибки при вычислении сил 0.1% и 1%, соответственно. А точность метода FMM зависит не только от количества членов в разложениях, но и от глубины иерархической структуры, которую необходимо увеличивать с ростом числа частиц в системе, этот эффект более детально исследовал Эзелинк [28]. На графике приводятся данные его работы для разложений 4-го, 7-го и 10-го порядков.
Кривые на рис. 7.4 демонстрирует несколько важных моментов: алгоритм TreeСode превосходит FMM в приложениях, где приемлема низкая точность вычисления сил ~0.1–1%, в которых он обгоняет прямой расчет на системах из 5000 частиц и более, тогда как FMM при той же точности делает это лишь на системах из 10000 частиц; однако если точность играет решающее значение, то FMM подтверждает свое преимущество над остальными методами, правда лишь на системах из 100000 частиц и более. Наша поточно-параллельная версия прямого расчета превосходит по скорости остальные методы вплоть до точки, соответствующей системам размером в 300000 частиц, давая при этом максимальную точность (в пределах машинной погрешности).
В настоящее время, широкое распространение различных технологий параллельных вычислений делает возможным моделирование достаточно больших систем даже с использованием прямого расчета с его квадратичной зависимостью времени счета от числа частиц, который является не только наиболее точным и простейшим из методов, но также реализует естественный параллелизм вычислений. На параллельных архитектурах с общей памятью достаточно распределить расчет сил отдельных частиц между процессорами, а для архитектур с распределенной памятью необходимо также применение того или иного способа минимизации издержек на передачу промежуточных результатов между процессорами.

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 10 11 |
