

Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
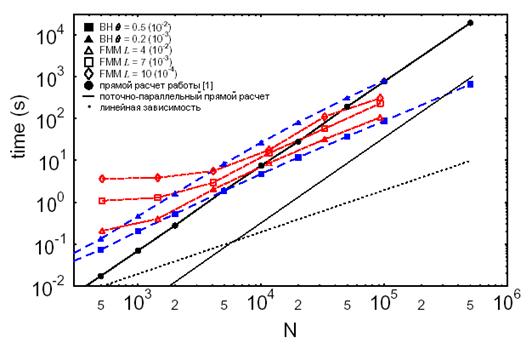
Рис. 7.4. Сравнение зависимости времени расчетов одного шага ММД от числа частиц в системе для разных методов по данным работы [17] и нашей поточно-параллельной методики.
Все вышеописанные «скоростные» алгоритмы разрабатывались для преодоления «квадратичного барьера» прямого расчета, однако, на практике параллельные версии этих методов реализуются с существенно различной эффективностью. Опишем основные стратегии распараллеливания каждого из них.
Параллельная версия классического метода Эвальда достаточно легко реализуема: во-первых, непосредственно разделяется расчет рядов в реальном и «обратном» пространстве; во-вторых, вычисление ряда в реальном пространстве может быть распределено между процессорами по аналогии с прямым расчетом; а ряд в «обратном» пространстве может быть преобразован в суммирование по частицам, умноженное на структурный фактор, которое также считается параллельно [29].
Распараллеливание сеточных методов, использующих быстрое преобразование Фурье (FFT) – довольно проблематично, т. к. бинарная рекурсивная природа FFT мало пригодна для реализации на архитектурах с распределенной памятью. Беккером была предложена альтернативная схема для решения уравнений поля на сетке [30], в которой вместо FFT используются итеративные методы, пригодные к распределенной параллельной реализации.
Реализация параллельных версий мультипольных алгоритмов требует наибольших дополнительных усилий, но привлекает потенциальной возможностью моделирования систем из 109 частиц на современных архитектурах с терафлоповой производительностью. Параллельная версия неадаптивного, двумерного FMM была предложена самим Грингардом в начале 90-х [31]. Эта схема, построенная на распараллеливании независимых стадий алгоритма, относительно эффективна на параллельных архитектурах с общей памятью, но мало пригодна для распределенных параллельных архитектур.
Из всех рассмотренных алгоритмов TreeCode Барнса-Хата, вероятно наиболее трудоемок в параллельной реализации. На параллельных архитектурах с общей памятью векторизация стадий построения дерева и создания списков взаимодействий позволяет добиться относительно неплохого прироста в скорости. Однако на распределенных архитектурах возникает трудно решаемая проблема – для построения списков взаимодействий необходим доступ ко всей структуре дерева, а поиск удаленных узлов дерева, находящихся в памяти других процессоров с использованием адресации указателями, которая типична для скалярных версий TreeCode, приводит к огромным коммуникационным издержкам. Сэлман и Уоррен [32] для решения этой проблемы изобрели практически новый алгоритм, отказавшись от указателей в пользу хэшированной схемы адресации.
В настоящее время, большая часть параллельных вычислительных экспериментов выполняется на кластерных архитектурах с распределенной памятью, так как они относительно эффективны во многих задачах и гораздо доступнее специализированных систем на архитектурах с общей памятью. Однако, как уже было отмечено выше, реализация многих «ускоренных» методов для моделирования систем с дальнодействием на кластерных архитектурах с распределенной памятью представляет определенные трудности. Поэтому в заключение этого обзора заметим следующее – выбор наилучшего алгоритма для конкретной задачи является открытым вопросом, и должен решаться исходя из анализа условий и целей моделирования и последующими экспериментальными исследованиями.
8.3. Методика высокоскоростного молекулярно-динамического моделирования диоксида урана
Для получения феноменологических характеристик кинетических процессов в вычислительных экспериментах необходимо длительное моделирование на системах с числом частиц, достаточно большим для получения статистической картины. Например, количество диффузионных прыжков при постоянной температуре прямо пропорционально количеству наблюдаемых частиц и времени наблюдения, однако, вычислительная нагрузка при моделировании систем с дальнодействием нарастает квадратично с увеличением числа частиц и линейно с увеличением времени моделирования, поэтому в таких исследованиях выгоднее увеличивать время наблюдения, а не число частиц. А значит важно добиться наименьших вычислительных затрат на один шаг времени.
Практика показала, что для моделирования диффузионных процессов достаточным является размер системы порядка 104 частиц. В частности, коэффициенты диффузии в системах с различными размерами в диапазоне 6144–20736 ионов отличались в пределах нескольких процентов. В результате экспериментального сравнения быстродействия различных подходов (прямой расчет, Ewald, PME, P3M, TreeCode, FMM) для систем такого размера выбор был сделан в пользу распараллеленного прямого расчета. Моделирование проводилось на специально разработанном программно-аппаратном комплексе на базе стандартной однопроцессорной рабочей станции (AMD Athlon1700+, 512 DDR pc3200, Nvidia nForce2 Ultra, Nvidia GeForce 6600GT), реализующем технологию поточно-параллельных вычислений. Динамика системы отображалась в реальном времени без ущерба производительности благодаря использованию аппаратного ускорения трехмерной графики с помощью технологии Microsoft Direct3D.
В качестве исходного приближения использовалась модель ионного кристалла, как с целочисленными, так и с дробными зарядами ионов. Взаимодействие между ионами описывали парными потенциалами взаимодействия вида:
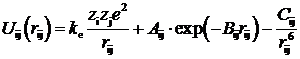
Здесь первый член описывает электростатическое кулоновское взаимодействие ионов, а второй и третий – силы отталкивания и дисперсионное притяжение электронных оболочек.
Таблица 7.1.
Параметры некулоновского потенциала для UO2 [33]
Параметр потенциала | A, эВ | B, Å | С, эВ×Å-6 |
U-O | 873.32735 | 2.477148 | 0 |
O-O | 50259.33984 | 6.542362 | 72.65339 |
В качестве параметров исходного некулоновского потенциала использовали данные из работы [33], модель “жестких” ионов (с целочисленными зарядами). Для дальнейшего улучшения свойств выбранного потенциала проводили уменьшение зарядов ионов до тех пор, пока значение параметра решетки диоксида урана при комнатной температуре ~300К не стало близким к экспериментальному, 5.47Å. Полученные таким образом значения заряда составили: для кислорода (-1.9085е), для урана (+3.817е).
Уравнения движения молекулярной динамики интегрировали конечно-разностным методом второго порядка точности – semi implicit Euler, в котором, в отличие от обычного метода Эйлера, расчет скоростей и позиций происходит не «одновременно», а «последовательно» – сначала, интегрируя силы, получают «новые» скорости, а затем на их основе вычисляют «новые» позиции:
vi(t+Δt/2) = vi(t-Δt/2) + Δt∙Fi(t)/mi
ri(t+Δt) = ri(t) + Δt∙vi(t+Δt/2)
Величину временного шага Δt варьировали в диапазоне от 3·10‑15с до 6·10‑15с.
Моделирование проводили в нулевых граничных условиях (НГУ). В вакууме формировали кубический нанокристалл, содержащий различное (от 2592 до 20736) число ионов, таким образом, чтобы его электростатический дипольный момент был минимальным. При этом кристалл в виде куба с ребром в K элементарных ячеек разбивали на три зоны: внешнюю (поверхность), промежуточную, с толщиной в одну элементарную ячейку каждая, а также внутреннюю, в виде куба с ребром в (K–4) элементарных ячейки. Динамические и структурные характеристики для каждой из них можно исследовать независимо. В частности, нанокристалл из 6144 ионов (K = 8) содержит: 768 частиц во внутренней, 1824 в промежуточной и 3552 во внешней областях. Для уменьшения влияния поверхности на расчеты объемных параметров последние рассчитывались только во внутренней зоне кристалла.
Коэффициенты диффузии ионов при заданной температуре рассчитывали по формуле Эйнштейна через зависимость среднеквадратичного смещения частиц <Δr2> от времени t:
D = <Δr2> / 6t
![]()
Среднеквадратичные смещения ионов рассчитывали следующим образом:
![]()
Здесь на шаге времени t идет суммирование по всем частицам сорта S квадратов смещений их текущих позиций относительно начальных.
8.4. Экспериментальные результаты и их обсуждение
Разработанный программно-аппаратный комплекс, реализующий технологию поточно-параллельных вычислений, позволил ускорить время расчетов на порядок по сравнению со специально оптимизированными версиями программ молекулярной динамики, использующими SIMD-технологии (SSE, 3Dnow!), и на два порядка по сравнению с простейшими реализациями из других работ [34] (табл. 7.2).
На его базе проводилось моделирование кристаллов UO2 с различным числом частиц (от 2592 до 20736). Рассчитаны коэффициенты диффузии собственных ионов (в том числе, для малоподвижного урана, процессы переноса которого методом молекулярной динамики практически не исследованы) в широком диапазоне температур, включающем все три состояния: кристалл, суперионное состояние и расплав. В результате моделирования удалось воспроизвести соответствующие фазовые переходы (суперионный и плавление) и рассчитать энергии активации диффузии для разных фазовых состояний.
С использованием новой методики стало доступно моделирование систем порядка 104 частиц на временах наблюдения порядка 106~108 шагов, что позволило обнаружить скачок коэффициента диффузии кислорода при суперионном переходе (рис. 7.5), полученные результаты находятся в согласии с имеющимися экспериментальными данными [35] и другими работами по молекулярно-динамическому моделированию [36] в пределах погрешностей эксперимента: энергия активации диффузии в кристаллической фазе составила 2.7735±0.2 эВ, а после суперионного перехода понизилась до 1.6823±0.2 эВ.
Таблица 7.2.
Сравнение скорости расчетов на кристалле из 6144 ионов
Программно-аппаратная реализация молекулярной динамики | Время, затрачиваемое на расчет | ||
1 шаг (сек) | 100.000 шагов (часы) | 1.000.000 шагов (сутки) | |
* Простейшая реализация из работы [34] (Delphi) | 15 | 416,7 | 173,6 |
* SIMD реализации авторов (С++, SSE, 3Dnow) и (С#, .NET) | 1 и 1,2 | 27,8 и 33,3 | 11,6 и 13,9 |
MPI распараллеленная реализация авторов (кластер) | 0,192 | 5,3 | 2,2 |
* поточно-параллельная реализация авторов (С#, asm) | 0,115 | 3,2 | 1,3 |
* все расчеты проводились на аппаратной базе рабочей станции – AMD Athlon1700+, 512 DDR PC3200, Nvidia nForce2 Ultra, Nvidia GeForce6600GT; кроме MPI версии, которая запускалась на кластере из 16 процессоров AMD Opteron.
8.5. Анализ зависимостей среднего квадрата смещений ионов кислорода от времени
Значения коэффициентов диффузии ионов урана и кислорода в модельных кристаллах при заданной температуре T, в соответствии с формулой, определяли по наклону зависимостей среднего квадрата смещений ионов от времени:
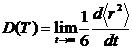 .
.
Проведенные расчеты показали, что при временах моделирования, порядка и бόльших 100 тыс. шагов ´ 5∙10-15 c (возрастающих с понижением температуры), зависимости <r2> от времени как для ионов кислорода, так и для ионов урана действительно в среднем выходят на прямые d<r2>/dt = Const, что подтверждает корректность определения коэффициентов диффузии.

Рис. 7.5. Характерная зависимость среднего квадрата смещений ионов кислорода от времени моделирования.
По вертикальной оси отложен средний квадрат смещений <r2>, умноженный на количество внутренних ионов O2-, по которым проводилось усреднение, так что высоты ступенек численно равны суммам квадратов перемещений ионов в новые равновесные позиции. A – зависимость <r2> от времени, полученная в численном эксперименте; B – прямая, наклон которой дает коэффициент самодиффузии кислорода при больших временах моделирования; 1 - 13 – ступеньки, соответствующие конкретным перемещениям ионов.
С другой стороны, оказалось, что при температурах, достаточно низких по сравнению с температурой плавления, начальные участки зависимостей <r2> от времени для ионов кислорода имеют ступенчатый характер, отслеживающий образование и перемещение единичных дефектов анионной подрешетки. Анализ ступенек позволил нам подтвердить диффузионный характер перемещения ионов, а также сделать некоторые выводы о механизмах собственного разупорядочения модельных кристаллов диоксида урана.
Характерная для сравнительно низких температур зависимость среднего квадрата смещения внутренних ионов кислорода в модельных кристаллитах от времени показана на рис. 7.5. Как отмечено выше, при обработке ступенек на этой и других аналогичных зависимостях мы считали, что каждая ступенька обусловлена перемещением одного или нескольких ионов кислорода из некоторых равновесных позиций в новые равновесные позиции, такие, из которых ионы не совершают дальнейших прыжков в течение временных интервалов, больших по сравнению с длиной ступеньки. Называть эти позиции равновесными возможно потому, что длины ступенек на порядки превышают времена гармонических колебаний ионов (около 10-13с., т. е., 20-ти шагов).
Для количественного анализа ступенек, мы сравнивали их высоты (нормированные, как показано на рис. 7.5) с квадратами переходов между равновесными позициями в анионной подрешетке диоксида урана, которая имеет простую кубическую структуру. Самые короткие из таких переходов показаны на рис. 7.6, их длины в единицах периода решетки приведены в табл. 7.3.
Как видно из рис. 7.6 и табл. 7.3, структура анионной подрешетки диоксида урана такова, что минимальные расстояния между соседними равновесными позициями ионов кислорода равны половине периода решетки (0.5a), а квадраты этих – расстояний r2=0.25a2. Расчеты показали, что минимальные достоверно разрешимые ступеньки действительно имеют высоты (0.25¸0.35)a2 (например, ступенька № 8 на рис. 7.5), которые могут соответствовать переходам анионов кислорода в соседний вакантный узел (переход 1 на рис. 7.6).
Следующие по размерам ступеньки (№ 1,3 на рис. 7.5) имеют высоты около 0.5a2 и могут соответствовать переходам ионов в вакантные позиции по диагоналям граней кислородной подрешетки (переход 2 на рис. 7.6), либо обменам позициями вдоль ребра между соседними ионами (парный переход 1 на рис. 7.6).
Далее, на на графике (рис. 7.5) есть ступеньки, имеющие высоты 1.05a2, 1.09a2 (№5,7 на рис. 7.5 и в табл. 7.5), которые могут соответствовать обмену позициями между соседними ионами по диагонали грани, либо четырем последовательным скачкам анионной вакансии вдоль ребер (что равносильно двум скачкам по диагоналям граней).
Образование устойчивых анионных вакансий было бы невозможно без перехода некоторых анионов в устойчивые междоузельные позиции (переходы 5,6). Таким переходам на рис. 7.5 могут соответствовать ступеньки №2,11, высотой 1.22a2 и 0.70a2.
На графике сравнительно низкие ступеньки, рассмотренные выше, чередуются со высокими, в несколько раз превосходящими a2. Можно предположить, что высокие ступеньки обусловлены быстрым перемещением высокоподвижных вакансий сразу на несколько позиций, до рекомбинации с междоузельным ионом, либо сложным коллективным движением нескольких ионов.

Рис. 7.6. Возможные диффузионные перемещения ионов кислорода в UO2. Показан фрагмент анионной плоскости системы [001]. Int – междоузельные позиции; эти позиции расположены в одной плоскости с катионами U4+, на 0.25∙a «выше», чем анионная плоскость. Квадраты длин переходов в единицах периода решетки a, под указанными номерами 1-5, приведены в табл. 7.3.
Таблица 7.3
Возможные диффузионные перемещения ионов кислорода в UO2
№ | Перемещение | D(N∙<r2>), a2 |
1 | Переход иона в соседний вакантный узел Обмен позициями между соседними ионами | 0.250 0.500 |
2 | Переход иона в соседний вакантный узел Обмен позициями между соседними ионами | 0.500 1.000 |
3 | Переход иона в соседний вакантный узел Обмен позициями между соседними ионами | 0.750 1.500 |
4 | Переход в ближайшую междоузельную позицию *) | 0.188 |
5 | Переход в удаленную междоузельную позицию | 0.688 |
6 | Переход в удаленную междоузельную позицию | 1.188 |
*) Переход в ближайшую междоузельную позицию неустойчив, поскольку ион может легко вернуться обратно |
Таблица 7.4
Высоты ступенек, полученных численным экспериментом при T = 2000K
№ | D(N∙<r2>), a2 | № | D(N∙<r2>), a2 |
1 | 0.57 | 7 | 1.05 |
2 | 1.22 | 8 | 0.26 |
3 | 0.57 | 9 | 3.80 |
4 | 2.75 | 10 | 3.01 |
5 | 1.09 | 11 | 0.70 |
6 | 3.10 | 12 | 2.71 |
С увеличением времени моделирования склоны ступенек из вертикальных становятся диагональными, а сами ступеньки постепенно выравниваются по высоте и сливаются в наклонную прямую. Этот процесс можно объяснить установлением равновесной (и неизменной) концентрации дефектов, обеспечивающих постоянный коэффициент диффузии. Видно, что при температурах, порядка и меньших 2500 K, равновесие наступает после нескольких сотен тысяч шагов, что показывает необходимость больших времен моделирования.
9. Восстановление потенциалов межчастичных взаимодействий по температурной зависимости периода решетки методами высокоскоростного МДМ на графических процессорах
9.1. Задача восстановления потенциалов межчастичных взаимодействий в кристаллах
Моделирование физических систем методами частиц (в частности, методом молекулярной динамики) предполагает описание взаимодействия этих частиц посредством некоторых модельных потенциалов взаимодействия (например, ). Точность моделирования в очень большой мере определяется адекватностью используемых потенциалов, которые в принципе можно либо рассчитывать из первых принципов (для чего обычно используют квантово-химические методы), либо восстанавливать из экспериментальных данных. Высокоскоростное поточно-параллельное моделирование можно эффективно применять как при квантово-химических расчётах, так и для поиска параметров потенциалов, обеспечивающих наилучшее совпадение свойств модели с экспериментом. Ниже мы рассмотрим решение задачи восстановления из экспериментальных данных парных потенциалов взаимодействия ионов в кристалле диоксида урана.
9.2. Исходные данные и метод восстановления потенциалов
В качестве опорных экспериментальных данных для восстановления параметров парных потенциалов взаимодействия (ПП) мы использовали зависимости теплового расширения периода кристаллической решетки различных типов АО-топлива при атмосферном давлении [37].
МД моделирование проводили в периодических граничных условиях (ПГУ) для исключения поверхностных эффектов; тогда объем модельного кристалла при данной температуре связан с его периодом решетки соотношением: V(T) = c*L(T)3, где с – количество элементарных ячеек в периодически-транслируемой кубической области.
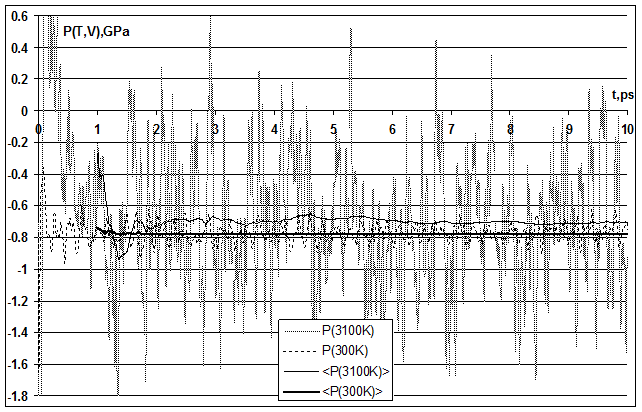
Искали параметры ПП, которые минимизируют среднеквадратичное отклонение периода решетки (или объема кристалла с заданным числом частиц) от экспериментального во всем диапазоне температур от комнатной до точки плавления 300~3100К при нулевом внутреннем давлении. Атмосферным давлением ~0.1 МПа пренебрегали в сравнении с давлениями 0.3~3 ГПа (см. рис. 1 и рис. 2), возникающими в кристалле при отклонениях периода решетки на 0.001~0.01 ангстрем (далее просто Å).
Для получения среднего периода решетки в «изобарном» МД-моделировании (при заданном числе частиц, давлении и температуре – NPT) требуется релаксация к равновесию и усреднение колебаний объема кристалла под действием баростата с периодом порядка 1-10 пикосекунд (далее просто пс), который определяется константой баростата. При этом из уравнения состояния следует, что отклонения объема при данной температуре однозначно определяются соответствующими отклонениями давления – к примеру, непосредственно из определения изотермической сжимаемости: dlnV = –BTdP.
Однако, для получения среднего давления в «изохорном» МД-моделировании (при заданном числе частиц, объеме и температуре – NVT) требуется релаксация к равновесию и усреднение лишь тепловых колебаний частиц около положений равновесия, период которых меньше на 1-2 порядка ~0.1 пс (см. табл. 8.1).
Таблица 8.1
Сравнение NVT и NPT типов МД-моделирования.
Тип МД-моделирования | NVT | NPT |
Тип усредняемых колебаний | Тепловые колебания частиц около положений равновесия | Колебания объема кристалла под действием баростата |
Период колебаний, пс | ~0.1 | ~1-10 (зависит от константы баростата) |
Время релаксации, пс | 2 | 25-250 |
Время усреднения, пс | 2-10 (увеличивается с ростом температуры) | 50-500 |
Общее время МД-моделирования на 1 точку температуры, пс | 4-12 | 75-750 |

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 10 11 |
