

Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Таблица 4
Возраст дебюта идиопатических эпилепсий (в годах)
Диагноз | Количество больных (абс.) | Возраст (M±σ) |
РЭ | 91 | 7,8±3,7 |
ДЗЭ Панайотопулоса | 12 | 5,6±4,4 |
ДЗЭ Гасто | 24 | 10,2±4,4 |
ЮАЭ | 27 | 11,6±3,3 |
ИГСП | 15 | 12,1±4,5 |
ДАЭ | 42 | 5,4±2,1 |
Синдром Дузе | 5 | 1,7±1,0 |
ЮМЭ | 26 | 12,5±3,4 |
Примечание: РЭ – роландическая эпилепсия, ДЗЭ – доброкачественная затылочная эпилепсия, ЮАЭ – юношеская абсансная эпилепсия, ИГСП – эпилепсия с изолированными генерализованными судорожными приступами, ДАЭ – детская абсансная эпилепсия, ЮМЭ – юношеская миоклоническая эпилепсия, М – среднее значение в годах, ) σ – стандартное отклонение (в годах)
Идиопатические эпилепсии характеризуются также определённым возрастом дебюта. Самый ранний дебют отмечен при эпилепсии с миоклонически-астатическими приступами (1,7±1 г.), позже других дебютировали генерализованные эпилепсии с вариабельным фенотипом – юношеская абсансная (11,6±3,3 г.), эпилепсия с изолированными генерализованными судорожными приступами (12,1±4,5 г.) и юношеская миоклоническая эпилепсия (12,5±3,4 г.) (таблица 4).
Эпилептические энцефалопатии.
Эпилептические энцефалопатии составили 5,22% случаев среди всех эпилепсий и эпилептических синдромов. Отмечено статистически значимое преобладание (р<0,05) синдрома Веста (67,39% или 3,51% случаев среди всех эпилепсий). Электрический эпилептический статус в фазу медленного сна встречался в 21,74% случаев (1,13% среди всех эпилепсий); частота синдрома Леннокса-Гасто составила 8,7% (или 0,45% среди всех эпилепсий). Синдром Отахара был выявлен у одного ребенка и составил 2,17% случаев среди эпилептических энцефалопатий (таблица 5).
Таблица 5
Структура эпилептических энцефалопатий (абс., %)
Форма | Количество больных | 95% доверительный интервал | % среди всех эпилепсий | 95% доверительный интервал | |||
Абс. | % | ||||||
Синдром Веста | 31 | 67,39* | 53,50 | 81,30 | 3,51 | 2,40 | 4,80 |
Синдром Леннокса-Гасто | 4 | 8,70 | 2,30 | 18,70 | 0,45 | 0,10 | 1,00 |
ЭЭСМ | 11 | 21,74^ | 10,90 | 35,00 | 1,13 | 0,50 | 1,90 |
Синдром Отахара | 1 | 2,17 | 0,00 | 8,50 | 0,11 | 0,00 | 0,70 |
Примечания: ЭЭСМ – электрический эпилептический статус в фазу медленного сна, * - статистически значимое преобладание синдрома Веста в сравнении со всеми формами (р<0,005), ^ - статистически значимое преобладание ЭЭСМ в сравнении с другими формами (р<0,005)
Среди вошедших в группу наблюдения с синдромом Веста было 14 (45,16%) мальчиков и 17 (54,84%) девочек. Заболевание дебютировало в возрасте до 5 месяцев у 20 (64,52%) пациентов и после 5 месяцев у 11 (35,48%) пациентов. Средний возраст дебюта составил 0,4±0,2 г. У большинства больных (93,55% - 29 человек) был диагностирован симптоматический синдром Веста и только у 6,45% (2 человека) - криптогенный.
Среди этиологических факторов симптоматического синдрома Веста в наблюдаемой группе детей преобладали перинатальные и постнатальные гипоксически-ишемические поражения головного мозга (65, 52%), отмечена статистически значимая разница при сравнении с другими этиологическими факторами (р<0,05). У 13,79% пациентов к развитию синдрома Веста привели перинатальные внутричерепные кровоизлияния, такой же процент случаев составили внутриутробные инфекции (герпетическая, цитомегаловирусная). Грубые нарушения эмбрионального развития, вызывающие эпилептогенное повреждение коры головного мозга по данным магнитно-резонансного исследования, такие как шизэнцефалия, пахигирия и микроцефалия, были выявлены у 6,90% пациентов
Нормальный неврологический статус до дебюта инфантильных спазмов отмечался у 2 детей с криптогенным синдромом Веста (100%) и у 5 (17,24%) - с симптоматической формой. В 82,76% (24 человека) случаев при симптоматическом синдроме Веста наблюдалась задержка психомоторного развития с раннего возраста, которая прогрессировала после дебюта заболевания. Среди неврологических нарушений были отмечены: мозжечковая атаксия - у 3 человек, у 3 – хореоатетоз, в 9 случаях наблюдалась гидро - и микроцефалия, в 14 – нарушение функции центрального двигательного нейрона в виде пирамидных нарушений, геми – и тетрапареза. Эти нарушения сочетались с патологией зрительного и глазодвигательных нервов в виде атрофии дисков зрительных нервов, нистагма, расходящегося и сходящегося косоглазия - у 10 больных (рисунок 1). При сравнении неврологических нарушений статистически значимой разницы не отмечено (р>0,05).
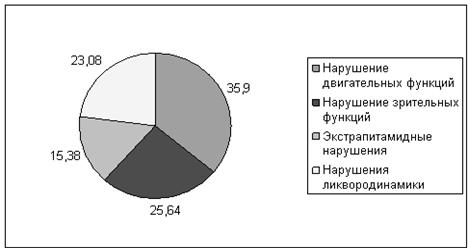

Рис.1. Неврологические нарушения при симптоматическом синдроме Веста (%)
Ядром клинической картины синдрома Веста являются эпилептические приступы в виде инфантильных спазмов, обычно приуроченные к пробуждению. У всех пациентов инфантильные спазмы группировались в кластеры от 4 до 20 спазмов сериями от 3 до 200 «кластерных атак» в сутки. Флексорные спазмы встречались у 18 (58%) больных, экстензорные – у 3 (10%), смешанные (флексорно–экстензорные) – у 10 (32%).
Электроэнцефалографическим эквивалентом инфальтильных спазмов является гипсаритмия – непрерывная генерализованная высокоамплитудная медленная и гиперсинхронная активность с острыми волнами, спайками и комплексами спайк–волна (типичная гипсаритмия). В исследуемой группе типичная (классическая) гипсаритмия была выявлена у 5 (16,12%) пациентов, из них у двоих детей с криптогенным и у троих с симптоматическим синдромом Веста. Различные виды атипичной гипсаритмии, характеризующиеся наличием локальных паттернов на фоне дизритмичной электроэнцефалограммы, а также эпизодами подавления биоэлектрической активности, наблюдались у 26 (83,88%) детей. Как видно на рисунке 2, у пациентов с атипичной гипсаритмией чаще встречались более грубые нарушения постнатального созревания нервной системы, сопровождающиеся задержкой психомоторного и речевого, а у детей с типичной гипсаритмией - психоречевого развития, но разница была статистически незначима (р=0,234).
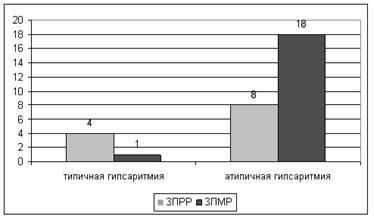
Рис. 2. Характер задержки развития в зависимости от типа гипсаритмии (абс. цифры)
Примечания: ЗПРР – задержка психоречевого развития. ЗПМР – задержка психомоторного и речевого развития.
Современные подходы к лечению синдрома Веста состоят в комбинации постоянного приема противоэпилептических препаратов в сочетании с курсовым назначением гормональной терапии. Применялась гормонотерапия синтетическим аналогом АКТГ синактеном-депо по схеме японских исследователей [Fukuyama, 1995] в более низких дозах для уменьшения побочного эффекта. Препарат вводился из расчета 0,0125 мг/кг на инъекцию по схеме: 2 недели ежедневно, 2 недели - через день, 2 недели – 2 раза в неделю, 2 недели -1 раз в неделю. Гормонотерапию получили 13 (41,94%) пациентов. На фоне терапии препаратом синактен депо длительная ремиссия приступов наступила в 6 (46,15%) случаях (у 1 ребенка с криптогенным и у 5 с симптоматическим синдромом Веста). У 4 (30,77%) пациентов зафиксировано уменьшение частоты приступов более чем на 50% от исходной, у 3 (23,08%) - незначительное снижение числа пароксизмов. В остальных случаях применялась базовая терапия препаратами вальпроевой кислоты в дозах от 30 до 60 мг/кг/сутки с последующим присоединением топирамата (3-5 мг/кг) у 6 человек. Ремиссия достигнута у 1 (7,14%) ребёнка, у 4 (28,57%) детей отмечено значительное уменьшение частоты приступов (более чем на 50% от исходной), у 9 (64,29%) детей частота инфантильных спазмов снизилась на 25%.
При катамнестическом наблюдении в сроки от 6 месяцев до 10 лет ремиссия приступов была зафиксирована у 10 (32,26%) пациентов, у 11 (35,48%) произошла трансформация в симптоматическую фокальную эпилепсию, у 5 (16,13%) - эволюция в синдром Леннокса-Гасто. У 4 (12,9%) детей раннего возраста сохранялся синдром Веста. 1 (3,23%) ребёнок умер от сопутствующей пневмонии (таблица 6).
Ремиссия наступила у всех детей с криптогенным синдромом Веста, а среди симптоматических форм только у больных с наименее тяжелыми поражениями, наличием типичной гипсаритмии на электроэнцефалограмме и рано начатым и интенсивным лечением с применением гормонотерапии и современных антиконвульсантов, но разница в частоте исходов среди больных получавших и не получавших гормонотерапию была статистически незначима (р>0,05).
Таблица 6
Исход синдрома Веста в зависимости от лечения (абс., %)
Исход | Количество больных | АКТГ (+) | АКТГ (-) | |||
Абс. | % | Кол-во больных абс. | % | Кол-во больных абс. | % | |
Ремиссия | 10 | 32,26 | 6 | 19,35 | 4 | 12,91 |
Эволюция в симптоматические фокальные эпилепсии | 11 | 35,48 | 1 | 3,22 | 10 | 32,26 |
Эволюция в синдром Леннокса-Гасто | 5 | 16,13 | 4 | 12,90 | 1 | 3,23 |
Текущий синдром Веста | 4 | 12,90 | 2 | 6,45 | 2 | 6,45 |
Смерть | 1 | 3,23 | 0 | 0,00 | 1 | 3,23 |
Общий исход | 31 | 100,00 | 13 | 41,93 | 18 | 58,07 |
Примечание: АКТГ (+) – больные получали гормонотерапию, АКТГ (-) – не получали гормонотерапию.

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 |
