
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral

Рисунок 4. Гетерофазная модель полимерного геля: 1 – полимерные цепи; 2 – полярные группы; 3 – вода ПР («связанная» вода); 4 – объем, занимаемый внешним раствором в геле; 5 – внешний раствор.
Так как в состав ПР входит полимер, то состав ПР и активности компонентов в нем отличаются от состава внешнего раствора, поэтому состав ПР отличается от состава раствора за его пределами. Другую часть объема геля занимает раствор, по своим свойствам не отличающийся от внешнего раствора (рис. 4). Объем фазы ПР определяют как сумму объемов самого полимера и воды, количество которой находят, используя ее изотерму сорбции. Если в фазе ПР присутствует растворенное вещество, то объем ПР состоит из объема полимера и объема раствора низкомолекулярного соединения, находящегося в ПР.
Как показано в [17], [18], объем, который при равновесии полимера с водой занимает в геле «свободная» вода практически не меняется при переносе данного полимера в растворы различной концентрации (рис. 5). Это свойство геля нарушается только в тех случаях, когда растворенное вещество проникает в фазу ПР. В остальных случаях, зная степень набухания геля и определив объем ПР, можно получить полную информацию о количестве воды и растворенного вещества в геле.
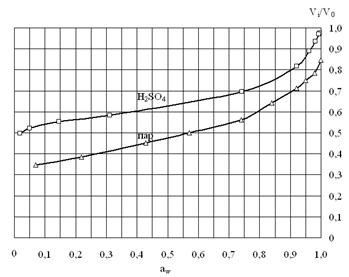
Рисунок 5. Влияние агрегатного состояния внешней фазы (пар – жидкость) на набухание сульфокатионита в Н-форме. V/V0 – относительный объем геля, где V0 – объём гранулы, набухшей в воде.
В тех случаях, когда растворенное вещество проникает в фазу ПР, его объем, также как и объем всего геля изменяются. В этом случае для расчета количества электролита в геле необходимо использовать способ, предложенный в работе [19].
3.2 Влияние количества сшивающего агента и противоиона на сорбцию воды
Известно, что в отсутствии химической реакции в случае межфазового равновесия химические потенциалы компонентов (m), присутствующих одновременно в обеих фазах, равны. В то же время их активности (a) обычно заметно различаются. Связь между активностями вещества, находящегося одновременно в обеих контактирующих фазах, обычно выражают через константы распределения ![]() этого вещества. Запишем
этого вещества. Запишем
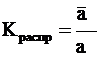 , (1)
, (1)
где ![]() – активность компонента в растворе сшитого полиэлектролита (РСПЭ), a – активность компонента в растворе низкомолекулярного электролита (РНМЭ).
– активность компонента в растворе сшитого полиэлектролита (РСПЭ), a – активность компонента в растворе низкомолекулярного электролита (РНМЭ).
Так как в рассматриваемой системе (вода – СПЭ – НМЭ) причиной образования второй фазы являются поперечные связи, то использование констант распределения компонентов позволяет через активности этих компонентов численно охарактеризовать влияние количества поперечных связей на количество и состав РСПЭ.
Появление инертных поперечных связей обычно не изменяет ни химические свойства, ни, тем более, природу полярных групп, но приводит к ситуации, в которой лимитируются расстояния между ними. Чем больше поперечных связей, тем меньше расстояние между группами в растворе СПЭ. Как результат: концентрация групп, а значит и активность воды в РСПЭ, становятся функциями количества поперечных связей. Для всех сшитых полиэлектролитов с заданным количеством поперечных связей существует предельная (максимальная) степень набухания в воде (при aw=1), значение которой не меняется с увеличением количества воды в системе вода – сшитый полиэлектролит.
В то же время изменение активности воды (aw) в равновесной с РСПЭ фазе приводит к изменению активности воды в полимере (![]() ), то есть к изменению концентрации РСПЭ и, соответственно, степени набухания СПЭ. Таким образом, становится очевидным, что существуют два параметра, влияющих на концентрацию РСПЭ, это – количество поперечных связей, которые определяют минимальную концентрацию РСПЭ и концентрация (точнее активность воды) в равновесном растворе НМЭ или паре. Суммарное влияние этих двух параметров на концентрацию РСПЭ иллюстрирует рис. 6. На нем хорошо видно влияние и активности воды во внешней фазе, и количества сшивающего агента на концентрацию РСПЭ.
), то есть к изменению концентрации РСПЭ и, соответственно, степени набухания СПЭ. Таким образом, становится очевидным, что существуют два параметра, влияющих на концентрацию РСПЭ, это – количество поперечных связей, которые определяют минимальную концентрацию РСПЭ и концентрация (точнее активность воды) в равновесном растворе НМЭ или паре. Суммарное влияние этих двух параметров на концентрацию РСПЭ иллюстрирует рис. 6. На нем хорошо видно влияние и активности воды во внешней фазе, и количества сшивающего агента на концентрацию РСПЭ.
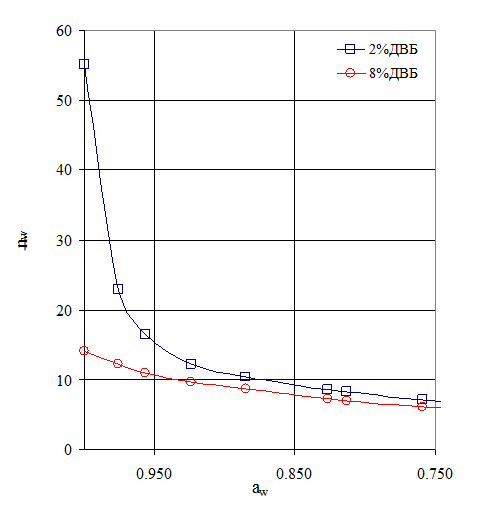
Рис. 6. Влияние активности воды и количества сшивающего агента на сорбцию воды полярными группами сульфокатионита в Н+–форме [13].
Чтобы найти численные значения констант распределения воды для полиэлектролита с разным количеством поперечных связей, воспользуемся данными по изучению сорбции воды из пара этим СПЭ и зависимостью nw = f(aw) для мономера, из которого он синтезирован (рис. 7).
Для бинарного раствора любого электролита каждому значению концентрации соответствует единственное значение активности воды. Как указывалось выше, свойства полярных групп СПЭ не отличаются от свойств этих групп в соответствующем НМЭ. Следовательно, при одинаковой концентрации полярных групп, то есть при ![]() = nw, активности воды в растворе СПЭ и в растворе НМЭ тоже будут одинаковы. Таким образом, равенство
= nw, активности воды в растворе СПЭ и в растворе НМЭ тоже будут одинаковы. Таким образом, равенство ![]() = nw в РСПЭ и в РНМЭ обеспечивает равенство активностей воды, из которого и находят активность воды (
= nw в РСПЭ и в РНМЭ обеспечивает равенство активностей воды, из которого и находят активность воды (![]() ) в РСПЭ.
) в РСПЭ.
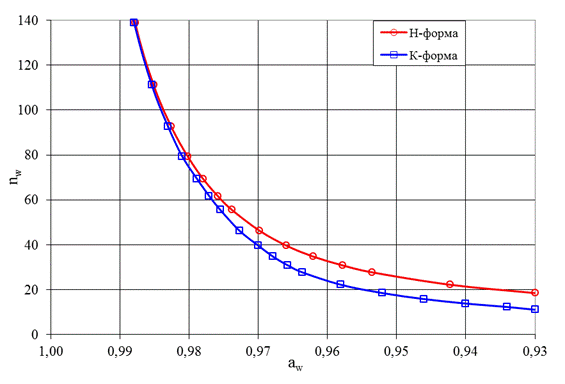
Рис. 7. Зависимость nw = f(aw) для бензолсульфокислоты и ее калиевой соли [20]
Находить константу распределения воды для конкретного СПЭ удобнее всего, воспользовавшись значением ![]() , которое соответствует максимальной сорбции воды при активности воды во внешней фазе аw = 1. Если величина
, которое соответствует максимальной сорбции воды при активности воды во внешней фазе аw = 1. Если величина ![]() известна, то нетрудно найти соответствующее ей значение aw. Оно будет характеризовать различие в активностях воды между фазами. То есть при активности воды во внешнем растворе, равной единице, активность воды в фазе РСПЭ будет равна
известна, то нетрудно найти соответствующее ей значение aw. Оно будет характеризовать различие в активностях воды между фазами. То есть при активности воды во внешнем растворе, равной единице, активность воды в фазе РСПЭ будет равна ![]() , а так как
, а так как
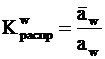 , (2)
, (2)
то найденное значение ![]() будет численно равно
будет численно равно ![]() потому, что aw = 1.
потому, что aw = 1.
Из сказанного выше следует, что на ![]() не влияют ни состав внешнего раствора НМЭ, ни природа противоиона полярной группы, а только природа полимера, а также природа и количество поперечных связей.
не влияют ни состав внешнего раствора НМЭ, ни природа противоиона полярной группы, а только природа полимера, а также природа и количество поперечных связей.
3.3 Методы, изучающие взаимодействие сшитых полиэлектролитов с водяным паром, водой и водными растворами
3.3.1 Изопиестический метод
При исследовании систем полимер – вода наиболее часто применяли изопиестический метод, разработанный в 1882 году Де Врие [21]. Его суть заключается в том, что при постоянной температуре в эксикаторы с известным давлением пара ![]() , где
, где ![]() – давление пара в эксикаторе,
– давление пара в эксикаторе, ![]() – давление пара над чистой водой, создаваемым или растворами серной кислоты разной концентрации или насыщенными растворами солей, помещали навески предварительно высушенного полимера. Навески периодически взвешивали. По достижении равновесия рассчитывали количество молей поглощенной воды, приходящихся на единицу массы сухого полимера. По результатам эксперимента строили изотерму сорбции воды данным полимером.
– давление пара над чистой водой, создаваемым или растворами серной кислоты разной концентрации или насыщенными растворами солей, помещали навески предварительно высушенного полимера. Навески периодически взвешивали. По достижении равновесия рассчитывали количество молей поглощенной воды, приходящихся на единицу массы сухого полимера. По результатам эксперимента строили изотерму сорбции воды данным полимером.
3.3.2 Метод динамической десорбционной порометрии
Подробное описание метода динамической десорбционной порометрии (ДДП) приведено в работе [22]. Метод основан на анализе кинетики сушки образца, проводимой в квазиравновесных условиях. Эти условия обеспечиваются ограничением скорости испарения пара растворителя из ячейки, в которой находится образец. Чем меньше скорость удаления пара из ячейки, тем ближе к равновесному значению давление пара в объеме ячейки.
Ячейку размещают на чашке термостатируемых весов и обдувают потоком сухого газа для гарантированного удаления паров воды с поверхности ячейки. В изотермических условиях через некоторое время в ячейке с образцом, пропитанным жидкостью, устанавливается квазистационарное распределение парциального давления пара жидкости, определяемое соотношением скоростей испарения жидкости из образца, массообмена внутри ячейки и удаления паров из ячейки. При относительно малой скорости испарения давление пара непосредственно в зоне испарения близко к равновесному давлению р, связанному с количествoм адсорбата в образце.
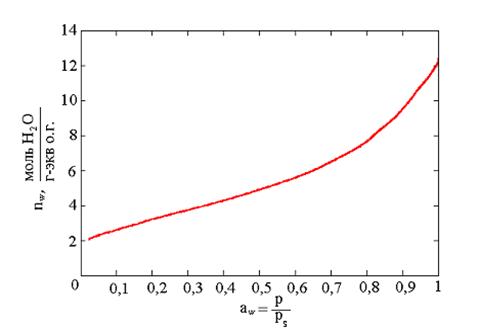
Рис. 8. Изотерма десорбции воды для катионита КУ – 2х8 в Н+-форме [23].
При данных условиях проведения процесса скорость испарения из ячейки будет простой функцией равновесного давления пара внутри ячейки над образцом р. Таким образом, оказывается возможным проводить изотермическую сушку образца, сохраняя над ним равновесное давление пара, со скоростью испарения, однозначно связанной с этим давлением. Это означает, что можно получить изотерму десорбции жидкости из анализируемого образца (рис. 8). Возможность использования данного метода для изучения воды в полимерах показана в работе [23].

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 10 11 |
