
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Из различных методов определения СЭ наиболее прямой и точный - измерение минимальной энергии фотоотрыва электрона от отрицательного иона.
Для большинства атомов присоединение электрона - экзотермический процесс. Наиболее высоким по абсолютной величине сродством к электрону обладают атомы галогенов в последовательности Cl > F > Вг > I. Энергии ионизации и сродство к электрону молекул определяют также, как это сделано для атома.
Лекция №6
ТЕОРЕТИЧЕСКИЕ МЕТОДЫ, ПРИМЕНЯЕМЫЕ ПРИ ИЗУЧЕНИИ СТРОЕНИЯ МОЛЕКУЛ И ХИМИЧЕСКОЙ СВЯЗИ
План лекции:
1. Молекула. Потенциальная поверхность. Равновесная конфигурация
2. Теория химической связи и её задачи.
3. Вариационный метод решения уравнения Шрёдингера.
§1. Молекула. Потенциальная поверхность. Равновесная конфгурация.
В химии молекулой называют наименьшую частицу данного вещества, обладающую его химическими свойствами, способную к самостоятельному существованию. Если отвлечься от поступательного движения молекулы как целого, то в ее энергию вносят вклад три вида движения: 1) движение электронов в поле ядер, 2) колебание ядер около положения равновесия и вращение молекулы вокруг оси, проходящей через центр масс, причем Еэл >> Екол >>Евр.
Хотя эти движения взаимосвязаны, можно приближенно рассматривать их как независимые и считать энергию молекулы равной сумме электронной, колебательной и вращательной энергий:
Е = Еэл + Екол +Евр. (6.1)
Волновая функция молекулы в этом приближении равна произведению функций, описывающих указанные три вида движения:
![]() (6.2)
(6.2)
Остановимся на наиболее важной составляющей энергии молекулы - электронной энергии. Так как скорость тяжелых ядер во много раз меньше скорости легких электронов, приближенно можно рассматривать движение электронов в молекуле в каждый данный момент, считая ядра неподвижными (приближение Борна - Оппенгеймера). Выбранному фиксированному положению ядер R отвечает определенная энергия электронов ![]() , включающая их кинетическую энергию, энергию взаимодействия электронов друг с другом и энергию взаимодействия электронов с ядрами.
, включающая их кинетическую энергию, энергию взаимодействия электронов друг с другом и энергию взаимодействия электронов с ядрами.
 Условимся включать сюда также энергию отталкивания ядер
Условимся включать сюда также энергию отталкивания ядер  . Тогда название "электронная" для
. Тогда название "электронная" для  указывает, что учитывается движение только электронов, но не ядер, а фиксированное расстояние между ядрами R рассматривается как параметр. Индекс "эл" при этом можно опустить (Eкол и Евр здесь не рассматриваются). Если расстояние между ядрами R изменится, изменится поле ядер, в котором движутся электроны, изменится и электронная энергия системы
указывает, что учитывается движение только электронов, но не ядер, а фиксированное расстояние между ядрами R рассматривается как параметр. Индекс "эл" при этом можно опустить (Eкол и Евр здесь не рассматриваются). Если расстояние между ядрами R изменится, изменится поле ядер, в котором движутся электроны, изменится и электронная энергия системы ![]() . В этом смысле электронная энергия есть функция межъядерного расстояния и по отношению к движению ядер играет роль потенциальной энергии. Вид функции
. В этом смысле электронная энергия есть функция межъядерного расстояния и по отношению к движению ядер играет роль потенциальной энергии. Вид функции ![]() для двухатомной молекулы АВ изображает кривая о рис. 5.1., называемая потенциальной кривой. Когда атомы А и В удалены на бесконечное расстояние, электронная энергия
для двухатомной молекулы АВ изображает кривая о рис. 5.1., называемая потенциальной кривой. Когда атомы А и В удалены на бесконечное расстояние, электронная энергия ![]() равна сумме электронных энергий невзаимодействующих атомов А и В в основном состоянии:
равна сумме электронных энергий невзаимодействующих атомов А и В в основном состоянии: ![]() .
.
При сближении ядер электронная энергия ![]() понижается (сила притяжения электронов к ядрам преобладает над силами отталкивания между ядрами и между электронами). Затем потенциальная кривая проходит через минимум при R = rе и при дальнейшем сближении ядер
понижается (сила притяжения электронов к ядрам преобладает над силами отталкивания между ядрами и между электронами). Затем потенциальная кривая проходит через минимум при R = rе и при дальнейшем сближении ядер ![]() возрастает, стремясь к бесконечности при R →0 (преобладает сила отталкивания). Межъядерное расстояние R=re, отвечающее минимуму потенциальной кривой, называется равновесным. При R=re равнодействующая всех сил притяжения и отталкивания равна нулю, молекула находится в устойчивом состоянии. Этому состоянию отвечает строго определенное значение электронной энергии молекулы. За нуль отсчета энергии можно принять, как и для атома, энергию невзаимодействующих электронов и ядер. Разность между суммой энергий невзаимодействующих атомов и Eэл. мол называют энергией химической связи или энергией диссоциации молекулы, отсчитанной от минимума потенциальной кривой, и обозначают De (глубина "потенциальной ямы"):
возрастает, стремясь к бесконечности при R →0 (преобладает сила отталкивания). Межъядерное расстояние R=re, отвечающее минимуму потенциальной кривой, называется равновесным. При R=re равнодействующая всех сил притяжения и отталкивания равна нулю, молекула находится в устойчивом состоянии. Этому состоянию отвечает строго определенное значение электронной энергии молекулы. За нуль отсчета энергии можно принять, как и для атома, энергию невзаимодействующих электронов и ядер. Разность между суммой энергий невзаимодействующих атомов и Eэл. мол называют энергией химической связи или энергией диссоциации молекулы, отсчитанной от минимума потенциальной кривой, и обозначают De (глубина "потенциальной ямы"):
. (6.3)
Данная потенциальная кривая соответствует классическим представлениям. Однако в нее надо внести поправки, так как равновесное состояние неосуществимо с точки зрения квантовой механики: в этом состоянии ядра неподвижны, значит, одновременно точно определены координата (R = rе) и импульс (р = 0), что противоречит соотношению неопределенностей Гейзенберга. Параметры rе и De (рис. 6.1) относятся таким образом к гипотетическому равновесному состоянию. В действительности даже и при 0К ядра не зафиксированы при R - rе, а совершают колебания около положения равновесия. Реальная энергия молекулы при этом выше, чем предполагалась, на величину энергии "нулевых колебаний" ![]() (рис.6.1). При нулевых колебаниях расстояние R между ядрами изменяется, в результате чего нулевой колебательный уровень характеризуется некоторым усредненным значением r0, которое из-за асимметрии потенциальной кривой незначительно, отличается от гипотетического равновесного расстояния rе=0,751·10-10 м и 0,741·10-10 м соответственно для Н2).
(рис.6.1). При нулевых колебаниях расстояние R между ядрами изменяется, в результате чего нулевой колебательный уровень характеризуется некоторым усредненным значением r0, которое из-за асимметрии потенциальной кривой незначительно, отличается от гипотетического равновесного расстояния rе=0,751·10-10 м и 0,741·10-10 м соответственно для Н2).
Определяемая на опыте энергия диссоциации молекулы D0 отсчитывается не от минимума потенциальной кривой как De, а от уровня нулевых колебаний (рис.и связана с De соотношением
![]() (6.4)
(6.4)
Энергия диссоциации D0, служит мерой прочности химической связи и определяется как изменение энергии в процессе - АВ = А + В при 0 К в идеально-газовом состоянии. Если специальных указаний нет, то понимается, что как молекула АВ, так и атомы А и В находятся в основном электронном состоянии. Это определение сохраняет силу и для многоатомных молекул. Например, для молекулы AmBn энергией диссоциации будет изменение энергии в процессе AmBn = mA + nB.
Потенциальная поверхность. Равновесная конфигурация. При описании потенциальной кривой вместо Eэл. мол обычно используют символ E(R) или Е. Для многоатомной молекулы Е является функцией уже не одной, а нескольких пространственных координат Rij. Например, потенциальная энергия молекулы ABC является функцией трех независимых координат - R1(A - В), R2(B - С) и угла о(АВС) или расстояний R1(A - В), R2(B - С) и R3(А - С).
Для линейной молекулы с фиксированным углом α- 180° эта функция изобразится поверхностью в трехмерном пространстве (потенциальная поверхность) Устойчивому состоянию молекулы отвечает минимальное значение ее энергии E(АВС) и определенное относительное расположение ядер в пространстве, называемое равновесной конфигурацией молекулы с параметрами rе(А - В) и rе(В - С). Глубина потенциальной ямы определяет энергию химической связи De и по формуле (5.4) энергию диссоциации молекулы D0.
Для более сложной молекулы, чем линейная ABC, равновесная конфигурация и энергия равновесного состояния определяются положением минимума на потенциальной поверхности в многомерном пространстве. Если потенциальная поверхность имеет два (или более) минимума, для молекулы возможны два изомера или более, отличающиеся параметрами равновесной конфигурации и энергией. Если минимума на потенциальной поверхности нет, данная система нестабильна, при любом расположении ядер она распадается на невзаимодействующие атомы.
Так же как и атом, молекулу можно перевести в возбужденные электронные состояния (энергия возбуждения Те), каждому из которых отвечает своя потенциальная поверхность или кривая (кривая б на рис. 6.1).
Рассмотрев потенциальную кривую (поверхность), можно дать еще одно определение молекулы молекула — физически устойчивая система, состоящая из двух (или более) ядер и определенного числа электронов, состояние которой описывается потенциальной кривой (поверхностью) с минимумом.
Говоря о физической устойчивости, понимают, что соединение атомов в молекулу сопровождается понижением энергии системы. Данным здесь определением охватываются кроме обычных молекул (Н2, СН4 и др.) также радикалы (СН, ОН, СН3 и др.) и молекулярные ионы (Н+, O2- и др.). Этому отвечает одинаковый подход теории строения к изучению перечисленных типов частиц. В тех случаях, когда молекулы одноатомны (благородные газы, пары металлов), сохраняет силу аналогичное определение для атома.
Равновесная конфигурация предполагает жесткую фиксацию всех межъядерных расстояний в молекуле. Однако реальная молекула не является жесткой системой. Вместе с тем у огромного большинства молекул амплитуды колебаний ядер весьма малы по сравнению с межъядерными расстояниями и можно, пренебрегая колебаниями, рассматривать молекулы как жесткие системы ("квазижесткие" или "квазитвердые" молекулы).
Равновесные конфигурации молекул принято относить к тем или иным точечным группам симметрии.
Двухатомные молекулы подразделяются на молекулы с одинаковыми ядрами, или гомонуклеарные (например, Н2), и с неодинаковыми ядрами, или гетеронуклеарные (например, HCI). Свойства симметрии их различны.
Симметрия равновесной конфигурации определяет и симметрию электронного облака молекулы. В связи с этим гомонуклеарные и гетеронуклеарные молекулы различаются по электрическим и оптическим свойствам, таким, как дипольный момент, поляризуемость и магнитная восприимчивость, правила отбора в спектрах. То же относится и к многоатомным молекулам, различающимся по симметрии, как, например, СН4 и СН3С1.
Рассмотренные молекулярные параметры: энергия диссоциации, межъядерные расстояния, равновесная конфигурация важны для химии не только как индивидуальные характеристики молекул. По ним можно рассчитать термодинамические свойства веществ и константы равновесия химических реакций.
§2. Теория химической связи и ее задачи. Уравнение Шредингера для молекул
Взаимодействие атомов, приводящее к образованию молекул простых и сложных веществ, а также кристаллов, называют химической связью. Взаимодействие атомов многообразно, поэтому многообразны и химические связи, которые часто сводят к нескольким основным типам: ковалентной, ионной, донорно-акцепторной, водородной связи и др. Однако все эти взаимодействия можно описать с позиций единой теории химической связи.
1) Эта теория призвана объяснить, какие силы действуют между атомами, как атомы объединяются в молекулы, что обеспечивает устойчивость образовавшейся сложной частицы (то же относится к кристаллам, жидкостям и другим телам).
2) Теория должна объяснить опытные факты, лежащие в основе классического понятия валентности, и наряду с этим существование и устойчивость многочисленных соединений, не укладывающихся в привычные рамки классических структурно-химических представлений.
3) Теория должна разработать единые методы расчета молекулярных параметров, интерпретировать молекулярные спектры.
4) Наконец, теория должна сделать возможным априорный расчет скорости химического процесса, зависимости ее от строения молекул реагирующих веществ.
 |
Современная теория химической связи, теория строения молекул и кристаллов базируется на квантовой механике: молекулы, как и атомы, построены из ядер и электронов, и теория химической связи должна учитывать корпускулярно-волновой дуализм микрочастиц. До применения методов квантовой механики к химии не удавалось создать непротиворечивую теорию химической связи.
Её фундамент был заложен в 1927 г. Гейтлером и Лондоном. Выполнив на основе квантовой механики расчет свойств молекулы водорода, они показали, что природа химической связи электрическая, никаких особых сил химического взаимодействия не существует. Действующие в молекуле между ядрами и электронами гравитационные и магнитные силы пренебрежимо малы по сравнению с электрическими силами.
Квантовомеханический подход к исследованию строения атома и молекулы один и тот же: нужно составить и решить уравнение Шредингера для системы из электронов и ядер и дать физическую интерпретацию полученным решениям. Составляя уравнение Шрёдингера для электронной энергии молекулы в приближении Борна - Оппенгеймера, считают ядра неподвижными.
Следовательно, электронная энергия для молекулы не зависит от координат ядер, а только от фиксированного расстояния R между ними (рис. 6.2). Во внимание принимаются лишь изменения координат электронов. Простейшая из молекул молекулярный ион ![]() содержит один электрон и два ядра. Для одного электрона в поле двух ядер (ион
содержит один электрон и два ядра. Для одного электрона в поле двух ядер (ион ![]() ) уравнение Шредингера имеет вид
) уравнение Шредингера имеет вид
![]() . (6.5)
. (6.5)
Для молекулярного иона ![]() потенциальная энергия
потенциальная энергия
 (6.6)
(6.6)
включает притяжение электрона 1 к ядрам А и В (первые два члена) и отталкивание ядер (ядра А и В у ![]() совершенно одинаковы - это протоны). Для иона
совершенно одинаковы - это протоны). Для иона ![]() с учетом (6.6) уравнение Шредингера принимает вид
с учетом (6.6) уравнение Шредингера принимает вид
 , (6.7)
, (6.7)
где Ψ - одноэлектронная волновая функция (собственная функция Шредингера для системы с одним электроном). Индексы "эл. мол." при E и Ψ - опущены для упрощения записи.
Состояние молекулы Н2 описывается уже двухэлектронной функцией Ψ, зависящей от координат двух электронов (см. рис. 6.2).
Многоэлектронными будут и волновые функции более сложных молекул. Уравнение Шредингера для молекулы Н2 имеет вид
![]() (6.8)
(6.8)
Индексы 1 и 2 при операторах Лапласа указывают, что волновая функция молекулы ![]() дифференцируется по координатам первого (
дифференцируется по координатам первого (![]() ) и второго (
) и второго (![]() ) электронов. Усложнится и выражение для потенциальной энергии:
) электронов. Усложнится и выражение для потенциальной энергии:
 (6.9)
(6.9)
В (6.9) первые четыре члена обозначают потенциальную энергию притяжения электронов 1 и 2 к ядрам А и В соответственно, пятый член - потенциальную энергию взаимного отталкивания электронов 1 и 2, последний член - энергию отталкивания ядер. Аналогично записывается уравнение Шредингера и для многоатомных молекул. В уравнениях (6.7) и (6.8) используется координатная молекулярная функция Ψ. Полная волновая функция молекулы Фмол, учитывающая и спин, должна удовлетворять принципу Паули антисимметрии волновых функций и строится в виде определителя.
Для молекулы, так же как и для атома, точное решение уравнения Шредингера возможно лишь для системы, содержащей один электрон - для молекулярного иона типа ![]() . Уже для молекулы Н2 в выражении (6.9) появляется член
. Уже для молекулы Н2 в выражении (6.9) появляется член  (энергия электронного отталкивания), зависящий от координат двух электронов, и точное решение невозможно. Однако принципиальный ответ о природе химической связи можно получить и при помощи приближенных методов решения волнового уравнения.
(энергия электронного отталкивания), зависящий от координат двух электронов, и точное решение невозможно. Однако принципиальный ответ о природе химической связи можно получить и при помощи приближенных методов решения волнового уравнения.
§3. Вариационный метод решения уравнения Шрёдингера
Одним из широко применяемых при рассмотрении теории химической связи приближенных методов решения уравнения Шредингера является вариационный метод. Здесь коротко излагается его сущность.
Уравнение Шредингера (1.1) может быть представлено в так называемой операторной форме. Для этого разделим все члены уравнения на ![]() и перегруппируем члены. Получим
и перегруппируем члены. Получим
 .
.
Сумма двух действий, производимых над функцией Ψ в левой части, дифференцирование и умножение, может быть записана с помощью оператора ![]() ,
,
![]()
который называют оператором энергии, оператором Гамильтона или гамильтонианом. Уравнение приобретает лаконичную форму:
(6.10)
По такому же принципу строится оператор Гамильтона для многоатомных систем, например, для молекулы Н2
![]() (6.11)
(6.11)
Умножим обе части уравнения Шредингера (6.10) на функцию Ψ*, комплексно сопряженную с волновой функцией
 . (6.12)
. (6.12)
Если Ψ — действительная функция, то Ψ*=-Ψ. (Левую сторону уравнения (6.12) нельзя записать аналогично правой в виде  , так как в отличие от E
, так как в отличие от E ![]() - оператор, а не число). Возьмем интеграл от обеих частей (5.12) по всему пространству, причем Е как постоянную величину вынесем за знак интеграла:
- оператор, а не число). Возьмем интеграл от обеих частей (5.12) по всему пространству, причем Е как постоянную величину вынесем за знак интеграла:
 .
.
Отсюда имеем
 . (6.13)
. (6.13)
Если функция Ψ нормирована, знаменатель обращается в единицу и
![]() . (6.14)
. (6.14)
Если известно точное выражение для Ψ, то энергия системы может быть рассчитана по формулам (6.13) или (6.14). Однако, обычно не известны ни Ψ ни Е либо неизвестна Ψ. Тогда для отыскания Ψ и Е можно воспользоваться вариационным принципом: подставив в (6.13) или (6.14) вместо истинной функции приближенную к ней так называемую пробную функцию Ψпробн, получим отвечающее ей значение Е. Оно обязательно будет не ниже энергии основного состояния системы E0
![]() . (6.15)
. (6.15)
Это положение строго доказывается. Волновая функция описывает распределение электронной плотности в системе (атоме, молекуле). Если бы пробная волновая функция привела к значениям E<E0, значит, она отвечала бы состоянию более устойчивому, чем осуществилось в системе. Но это невозможно, так как электроны и ядра в атомах или молекулах, предоставленные самим себе, осуществляют состояние с наименьшей энергией Е0. Поэтому любая Ψпробн отличная от истинной, приведет при подстановке в (6.13) к значениям большим, чем Е0. Только если Ψпробн совпадает с истинной, она приведет к истинному значению Е0. т. е. к минимальному.
Задача сводится, таким образом, к нахождению минимума Е в уравнении (6.13). Обычно используется метод линейных комбинаций. Здесь пробная функция выбирается в виде линейной комбинации линейно-независимых базисных функций f (лучше всего ортогональных или ортонормированных) с независимыми параметрами с1 с2,..., сn, например атомных волновых функций или функций Слейтера:
Ψпробн = c1.f1 + c2.f2 +…+ cn. fn. (6.16)
Параметры с1 с2,..., сn неизвестны, их можно варьировать, добиваясь такого значения Ψпробн при котором выражение (5.13) достигает минимума. Условие минимума при наличии п независимых параметров в (5.16) приводит к системе из n уравнений и дает не одно значение Е, а п значений Е1, Е2, ..., Еп и отвечающие им п взаимно ортогональных волновых функций Ψ1, Ψ2, Ψ3,… Ψn.
Каждой Ψi отвечает свой набор параметров сi. Самое низкое из значений энергии Е1 наиболее близко к истинному значению энергии основного состояния, а Ψi к истинной волновой функции. В данном решении они отвечают основному состоянию. Остальные Ei и Ψi относятся к более высоким, возбужденным состояниям.
Увеличивая число слагаемых в (5.16), принципиально можно при помощи ЭВМ получить значения Ψ и соответствующие им Е, сколь угодно близкие к истинным. Однако число базисных функций нельзя увеличивать до бесконечности, работают с конечным базисным набором, который приводит к приближенным решениям.
Лекция №7
МЕТОДЫ, ПРИМЕНЯЕМЫЕ ПРИ ИЗУЧЕНИИ СТРОЕНИЯ МОЛЕКУЛ И ХИМИЧЕСКОЙ СВЯЗИ.
План лекции:
1. Метод валентных связей и метод молекулярных орбиталей
2. Приближённое описание молекулярной орбитали в методе МО ЛКАО.
3. Расчёт энергии и волновой функции ![]() в методе МО ЛКАО
в методе МО ЛКАО
§ 1. Метод валентных связей и метод молекулярных орбиталей
Функция Гейтлера - Лондона для молекулы ![]() - Работа Гейтлера и Лондона (1927) была основополагающей в области применения квантовой механики к химии, т. е. в области теории строения молекул. Эти ученые впервые нашли приближенное решение уравнения Шредингера для молекулы Н2, подойдя к ней как к системе, состоящей из двух атомов водорода. Использованная ими приближенная функция для молекулы H2 строилась из 1s атомных орбиталей каждого атома водорода. В нулевом приближении она имела вид, аналогичный функции для атома гелия:
- Работа Гейтлера и Лондона (1927) была основополагающей в области применения квантовой механики к химии, т. е. в области теории строения молекул. Эти ученые впервые нашли приближенное решение уравнения Шредингера для молекулы Н2, подойдя к ней как к системе, состоящей из двух атомов водорода. Использованная ими приближенная функция для молекулы H2 строилась из 1s атомных орбиталей каждого атома водорода. В нулевом приближении она имела вид, аналогичный функции для атома гелия:
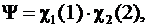 (A)
(A)
где ![]() атомная орбиталь электрона 1 в поле первого ядра;
атомная орбиталь электрона 1 в поле первого ядра; ![]() — атомная орбиталь электрона 2 в поле второго ядра. Величина
— атомная орбиталь электрона 2 в поле второго ядра. Величина ![]() определяет таким образом вероятность того, что электрон 1 находится у первого ядра и одновременно электрон 2 у второго, выражение (А) - частное решение уравнения Шредингера для H2. Однако в силу тождественности и неразличимости электронов возможен обмен электронами между ядрами, приводящий к состоянию той же энергии с функцией, являющейся вторым частным решением
определяет таким образом вероятность того, что электрон 1 находится у первого ядра и одновременно электрон 2 у второго, выражение (А) - частное решение уравнения Шредингера для H2. Однако в силу тождественности и неразличимости электронов возможен обмен электронами между ядрами, приводящий к состоянию той же энергии с функцией, являющейся вторым частным решением
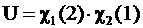 , (Б)
, (Б)
(так называемое обменное вырождение). В результате этого общая функция нулевого приближения (функция Гейтлера — Лондона) должна быть линейной комбинацией функций (А) и (Б):
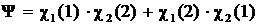 (7.1)
(7.1)
Используя функцию (6.1) Гейтлер и Лондон рассчитали потенциальную кривую молекулы H2 и нашли, что её минимуму отвечают энергия диссоциации Dе(H2) = 3,2 эВ и межъядерное расстояние rе(Н2) = 0м (экспериментальные значения Dе(Н2) = 4,75 эВ, re(H2) = 0,740-10 м).
Учитывая грубость использованного приближения для волновой функции, результаты надо считать вполне удовлетворительными. Значение этой работы чрезвычайно велико.
Во-первых, Гейтлер и Лондон показали, что уравнение Шредингера справедливо не только для атома, но и для молекулы, т. е. является фундаментальным.
Во-вторых, было показано, что химическая связь имеет электрическую природу, поскольку в уравнении Шредингера в качестве потенциальной энергии рассматривалась только энергия электростатического взаимодействия ядер и электронов [см. уравнение (6.9)], а результаты расчета вполне согласуются с опытом.
Как показали Гейтлер и Лондон, из (7.1) следует, что электронная плотность в области между ядрами в молекуле Н2 оказывается выше, чем простое наложение электронной плотности атомов. Эта повышенная плотность электронного заряда между ядрами удерживает их вместе, поскольку пребывание двух электронов в поле двух ядер энергетически выгоднее нахождения каждого из них в поле одного ядра. Пара электронов, ставшая общей двум ядрам, обусловливает химическую связь в молекуле. Так как функция (7.1) является симметричной, то из принципа Паули следует, что образование молекулы Н2 с такой функцией возможно только, если спины электронов антипараллельны. Полная волновая функция Ψмол будет при этом антисимметричной по отношению к перестановка координат электронов.
Если два атома имеют электроны с параллельными спинами, то система должна описываться другой координатной волновой функцией:
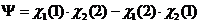 (7.2)
(7.2)
и при этом электронная плотность между ядрами понижена, вследствие чего возникает отталкивательное состояние, молекула Н2 не образуется. Обмен электронами с параллельными спинами приводит к отталкиванию - обменное отталкивание. Обмен электронами с антипараллельными спинами приводит к притяжению. Употребляют выражение "обменные силы" отталкивания и притяжения. Надо помнить об условности таких названий, так как химическая связь - результат электрического взаимодействия. Никаких новых обменных сил не возникает.
При разработке теории строения молекул в начале 30-х годов возникли и затем развивались два метода: 1) метод валентных связей. ВС-метод (разрабатывался Полингом, Слейтером и другими на основе работы Гейтлера и Лондона) и 2) метод молекулярных орбиталей МО-метод (развивался Малликеном, Хундом, Хюккелем и др.)
В высших своих приближениях они приводят к практически одинаковым результатам, достигаемым, однако, разной ценой. В более простом приближении каждый из них обладает преимуществами в описании одних явлений и недостатками при описании других.
В ВС-методе полагают, что каждая молекула составлена из атомов, и для объяснения электронного строения молекулы применяют атомные орбитали составляющих ее атомов. Подобное приближение диаметрально противоположно МО-методу, рассматривающему в своей наиболее общей форме каждую молекулу как самостоятельное целое, а не простую совокупность атомов.
ВС-метод. В методе валентных связей результаты работы Гейтлера и Лондона обобщены и распространены на многоатомные молекулы. Поэтому характерные особенности двухэлектронной связи в молекуле Н2 перенесены на связи в многоатомных молекулах типа СН4 и др.
1. Химическая связь образуется парой электронов с противоположными спинами, локализованной между двумя атомами (теория локализованных пар).
2. Число связей, образуемых данным атомом с другими атомами (валентность), равно числу неспаренных электронов внешней электронной оболочки в основном или в возбужденном состоянии.
3. Направленность валентности определяется ортогональностью атомных орбиталей центрального атома, участвующего в образовании связи, например, рх. и ру. АО кислорода взаимно перпендикулярны.
4. Аддитивность энергии связи и других свойств является следствием локализации пары электронов между двумя атомами, образующими связь: молекула рассматривается как сумма отдельных связей, а отсюда и свойства молекулы - сумма свойств связей.
5. Валентно-насыщенные молекулы могут образовать между собой химические соединения за счет донорно-акцепторного взаимодействия. Такое взаимодействие обусловлено наличием вакантной АО в одном из атомов акцептора и неподеленной пары электронов на АО одного из атомов молекулы донора. Донорно-акцепторная связь по своей природе является ковалентной, так как осуществляется парой электронов, общей двум атомам.
Для лучшего соответствия опытным данным необходимо ввести допущения об участии в химической связи атомов в возбужденном состоянии и о гибридизации атомных орбиталей. Изложенная здесь концепция метода валентных связей обладает определенной стройностью и наглядностью. Она приемлема для химика, так как переводит на язык квантовых представлений привычные структурные формулы, соотнося каждый валентный штрих в структурной формуле локализованной паре электронов. Это представление о локализованной паре электронов является квантовомеханическим аналогом более ранней идеи Льюиса о связи как о паре электронов, общей для двух атомов.
Однако метод валентных связей в простом приближении теории локализованных пар и направленных валентностей оказывается недостаточным для понимания структуры и свойств более сложных соединений.
Остановимся на проблемах, которые нельзя решить в простом приближении локализованных пар.
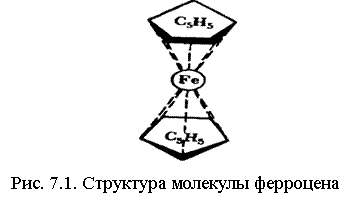
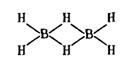
К ним относятся проблемы химических соединений благородных газов с галогенами (например, XeF6, XeOF2), структура сэндвичевых соединений, таких, как ферроцен (рис. 6.1), геометрия равновесных конфигураций ряда молекул, например различие в конфигурациях Li2О и Н2O. Сюда же относится проблема молекул с дефицитом электронов, когда число валентных электронов недостаточно для образования существующих связей, например, в диборане В2Н6,
 |
где 12 электронов обеспечивают по схеме парных взаимодействий 8 связей.
 |
Метод локализованных пар (простейшее приближение метода ВС) неудобен для описания огромного числа молекул, содержащих цепочки так называемых сопряженных связей С = С – С = С - С=, и молекул ароматических соединений, в том числе бензола и его производных. Эти соединения не могут быть описаны единственной структурной формулой. Уже молекуле бензола можно приписать, по меньшей мере, две структурные формулы Кекуле:
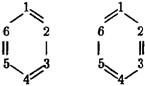
Так как валентный штрих в методе локализованных пар сопоставляется с локализованной парой электронов, то приведенным двум формулам Кекуле соответствуют, по меньшей мере, два разных распределения электронной плотности. Но для молекулы в стационарном состоянии существует одно единственное распределение. Поэтому в методе валентных связей реальное распределение электронной плотности молекулы бензола надо представить как суперпозицию, по меньшей мере, двух распределений локализованных пар, а для более точной картины - пяти распределений. Это значительно усложняет метод, не облегчая восприятия реальности. Для более сложных молекул число используемых при их описании валентных схем стремительно возрастает.
Метод полностью утрачивает преимущества наглядности, а в расчете молекулярных свойств предпочтительным является метод молекулярных орбиталей.
Метод молекулярных орбиталей. С 50-х годов метод МО занимает лидирующее положение. В методе МО не только значительно проще объясняются электронное строение и свойства сопряженных и ароматических соединений. Малликену, метод МО обладает рядом преимуществ, особенно в том случае, когда необходимы пространные теоретические расчеты, возможности которых постоянно возрастают с применением мощных вычислительных машин. Кроме того, метод МО значительно лучше приспособлен для исследования спектров и, следовательно, фотохимических свойств молекул, т. е. приспособлен к тому кругу вопросов, который в настоящее время пользуется все возрастающим пониманием.
Основные идеи метода молекулярных орбиталей.
Природа электронов в атомах и молекулах, взаимодействие их с ядрами и друг с другом в принципе те же, что и в атоме. Поэтому логично пользоваться для молекулы той же физической моделью, что и для атомов, - орбитальным приближением. Главные черты орбитального приближения в методе МО следующие:
1. Молекула рассматривается как целое, а не как совокупность сохраняющих некоторую индивидуальность атомов. Каждый электрон принадлежит молекуле в целом и движется в поле всех ее ядер и электронов.
2. Состояние электрона описывается одноэлектронной волновой функцией Ψ, характеризуемой определенным набором квантовых чисел. Функция эта называется молекулярной орбиталью (МО). В отличие от одноцентровой атомной орбитали (АО) молекулярная орбиталь в общем случае многоцентровая, так как число ядер в молекуле не менее двух. Как и для электрона в атоме, квадрат волновой функции ![]() определяет плотность вероятности нахождения электрона или плотность электронного облака.
определяет плотность вероятности нахождения электрона или плотность электронного облака.
Полное описание состояния электрона дает молекулярная спин-орбиталь, выражаемая как произведение МО на спиновую функцию:
3. Каждой МО соответствует определенная энергия Ei. Приближенно эта энергия равна энергии ионизации с данной орбитали (теорема Купманса). Орбитальная энергия слагается из кинетической энергии электрона, потенциальной энергии притяжения электрона ко всем ядрам и усредненной потенциальной энергии отталкивания электрона на МО от всех остальных электронов.
4. Совокупность МО молекулы, занятых электронами, будем называть её электронной конфигурацией.
Электронная конфигурация молекулы, так же как и для атома, строится на основе двух фундаментальных положений - принципа наименьшей энергии (электрон занимает в молекуле свободную орбиталь с наименьшей энергией) и принципа Паули (на одной МО не может находиться более двух электронов, при этом спины электронов должны быть антипараллельны). Следовательно, для описания электронной конфигурации основного состояния молекулы с 2n электронами (или 2п — 1) требуется n молекулярных орбиталей.
Вырожденные орбитали заполняются в соответствии с первым правилом Хунда. Электронные оболочки молекул, в которых на каждой заселенной орбитали находятся два электрона с антипараллельными спинами, называют закрытыми; при наличии хотя бы одной МО, заселенной неспаренным электроном, открытыми.

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 |
