
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Молекула H2
Электронная конфигурация молекулы Н2 в основном состоянии H2[(σgls)2]. В основном состоянии молекулы два ее электрона согласно принципу наименьшей энергии занимают наиболее низкую орбиталь σ1s и согласно запрету Паули имеют противоположные спины. Суммарный спин равен нулю, молекула является диамагнитной, мультиплетность 2s+1=1 - (синглет). Оба электрона занимают четную (g) орбиталь. По правилу произведения (g∙g = g) состояние системы четное. Схема заселенности МО молекулы Н2 и других молекул элементов первого периода приведена на рис. 7.2.
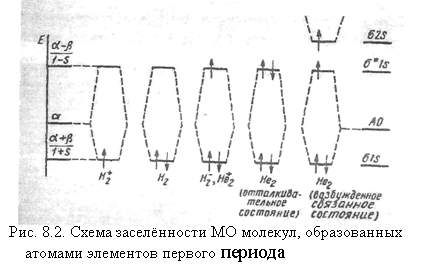
В многоэлектронных молекулах согласно орбитальному приближению полная электронная энергия = (сумма орбитальных энергий) - (усредненная энергия отталкивания электронов) + (энергия отталкивания ядер). Можно принять (Хюккель, 1931), что последние два члена компенсируют друг друга. В таком приближении полная электронная энергия равна сумме орбитальных энергий: ![]()
Соответственно энергия диссоциации равна
De(H2) = 2Е(Е) - E(Н2) = 2α-2(α+β) =-2β
Связь в Н2 прочнее, чем в ионе ![]() , вследствие того, что на
, вследствие того, что на ![]() -связывающей орбитали находятся два электрона вместо одного.
-связывающей орбитали находятся два электрона вместо одного.
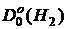 =4,478 эВ и
=4,478 эВ и 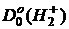 = 2,651 эВ, rе(Н2) = 0.7414Å и
= 2,651 эВ, rе(Н2) = 0.7414Å и ![]() =1,06 Å.
=1,06 Å.
Волновая функция. Координатная волновая функция основного состояния молекулы Н2 в нулевом приближении может быть представлена произведением одноэлектронных волновых функций
![]() , (8.2)
, (8.2)
где χ1 и χ2 — атомные орбитали при ядрах А и В; (1) и (2) — координаты первого и второго электронов.
В соответствии с принципом Паули полная волновая функция с учетом спина должна быть антисимметричной по отношению к перестановке координат электронов. А так как координатная функция (7.2) симметрична, то антисимметричной будет спиновая функция. А это значит, что на σ1s-MO два электрона должны иметь антипараллельные спины.
В первом возбужденном состоянии молекулы H2 один из электронов переходит на разрыхляющую σ*ls-MO. Координатная волновая функция
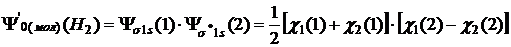
антисимметрична как произведение симметричной и антисимметричной МО. Следовательно, спиновая волновая функция должна быть симметричной (спины электронов параллельны). Суммарный спин S = 1.
Мультиплетность 2S+1=3 - (триплет), т. е. это состояние может осуществляться тремя способами, при которых Ms = 1; 0 и —1. Энергия системы Е = ЕS + ЕA ≈ (α + β) + (α—β) = 2α или, точнее, ![]()
т. е. выше, чем энергия двух электронов на АО, и связь не возникает; электрон на антисвязывающей МО разрыхляет связь сильнее, чем укрепляет ее электрон на связывающей. Состояние является отталкивательным, потенциальная кривая не имеет минимума. При встрече двух атомов водорода состояние отталкивания возникает в три раза чаще, чем состояние устойчивой молекулы, так как первое реализуется тремя способами, а второе – единственным.
Лекция№9
Теория химической связи
План лекции:
1. Гетеронуклеарные двухатомные молекулы
2. Полярная связь. Электрический дипольный момент молекулы
3. Донорно-акцепторная связь
§1. Гетеронуклеарные двухатомные молекулы
Гетеронуклеарными называют молекулы, содержащие ядра атомов разных элементов, например, СО, NO, HF, LiF и др.
Рассмотрим электронное cтроение молекулы CO. Электронные конфигурации атомов: C[1s22s22p2] и O[1s22s22p4]. Межъядерную ось примем за ось z. Ядра С и О имеют неодинаковые заряды (6 и 8). Поэтому соответствующие квантовые уровни атома кислорода лежат ниже, чем в атоме углерода (рис.8.1).
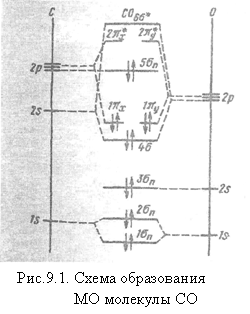
Поскольку в гетеронуклеарных молекулах в одной МО участвуют атомные функции разных состояний, например, 2s-АО углерода и 2p - АО кислорода, прежняя система записи конфигурации невозможна, и орбитали просто нумеруются по порядку. Так, запись конфигурации СО обычно принимает вид: ![]() .
.
Как видно из рис. 8.3, орбитали K-слоя 1σn и 2σn имеют ту же энергию, что и в атоме, так же как и низколежащая орбиталь 3σn. Индекс n указывает на несвязывающий характер орбиталей. Далее следуют связывающие орбитали 4σ, 1πx, образованные из 2s(С)- 2рx(О)- и 2py (O)- орбиталей. Несвязывающая орбиталь 5σn образована из 2px(C) орбитали и близка к ней по своей симметрии.
Перечисленные семь орбиталей заполнены 14 электронами молекулы СО. Разрыхляющие 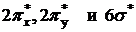 свободны.
свободны.
При таком рассмотрении связь оказывается тройной за счет 4σ21π4|-электронов. Такое представление отвечает очень высокой энергии диссоциации Dо(СО) = 11,11 эВ (это максимальное значение энергии диссоциации, наблюдаемое у двухатомных молекул) и весьма малому межядерному расстоянию re(СО) = 1,1222∙10-10 м (1,1222 Å). Обе эти молекулярные константы близки к константам для изоэлектронной молекулы N2.
Но в отличие от молекулы N2 верхняя занятая ![]() -орбиталь — несвязывающая. Электронная пара на такой орбитали называется неподеленной или уединенной парой (здесь — на углероде), что подчеркивает ее атомный характер. В отличие от неподеленной пары на кислороде (несвязывающая
-орбиталь — несвязывающая. Электронная пара на такой орбитали называется неподеленной или уединенной парой (здесь — на углероде), что подчеркивает ее атомный характер. В отличие от неподеленной пары на кислороде (несвязывающая ![]() -орбиталь), лежащей очень низко и поэтому нереакционноспособной, неподеленная пара
-орбиталь), лежащей очень низко и поэтому нереакционноспособной, неподеленная пара ![]() на углероде играет большую роль в химических реакциях оксида углерода. Она находится на орбитали, вытянутой в сторону, противоположную атому кислорода, и самой высокой по энергии. Благодаря этому СО является хорошим донором электронов. Незанятые орбитали молекулы СО именно
на углероде играет большую роль в химических реакциях оксида углерода. Она находится на орбитали, вытянутой в сторону, противоположную атому кислорода, и самой высокой по энергии. Благодаря этому СО является хорошим донором электронов. Незанятые орбитали молекулы СО именно ![]() — разрыхляющие, играют важную роль при образовании карбонилов переходных металлов.
— разрыхляющие, играют важную роль при образовании карбонилов переходных металлов.
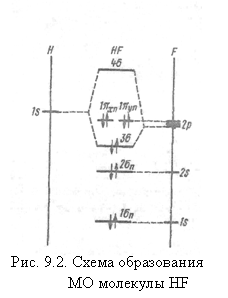
Примером гетеронуклеарных двухатомных молекул с ядрами, сильно отличающимися по величине эффективного заряда, могут служить молекулы гидридов. Рассмотрим молекулу HF. Электронные конфигурации атомов: H[ls], F[ls22s22p5]. Энергии ls-AO (H) и 2р-А0 (F) близки, и связывающая σ-орбиталь может быть представлена как линейная комбинация 1s-орбитали атома водорода и 2рz-орбитали атома фтора, имеющих одинаковые свойства симметрии относительно оси молекулы:
![]()
Различие в коэффициентах с1 и с2 отражает несимметричное распределение электронной плотности на МО. Упрощая, можно считать, что все электроны фтора, кроме 2pz, сохраняют свой атомный характер: ls-и 2s-орбитали не комбинируют с ls-орбиталью атома Н вследствие большого отличия от нее по энергии. АО 2рх и 2ру не комбинируют из-за различия по симметрии относительно оси молекулы. Все эти орбитали становятся несвязывающими МО. Основной вклад в химическую связь в молекуле HF вносит пара электронов на а-связывающей молекулярной орбитали.
Электронная конфигурация молекулы может быть записана в виде
![]()
или 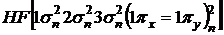
§ 2. Полярная связь. Электрический дипольный момент молекулы
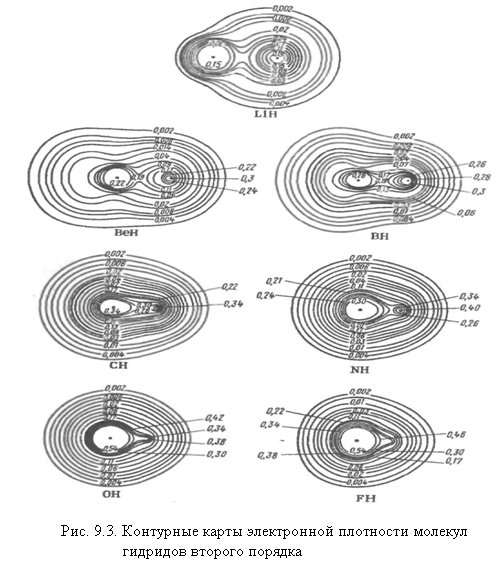
Для всех гетеронуклеарных молекул можно отметить характерную особенность: электронная плотность в них распределена несимметрично относительно обоих ядер (рис. 9.3). При таком распределении электронной плотности химическую связь называют полярной или точнее полярной ковалентной связью, а молекулы полярными. У молекул гидридов у HF особенно заметно проявляется несимметричное распределение заряда.
Электронная плотность вследствие большого различия в эффективных зарядах водорода и фтора смещена в сторону последнего. Вследствие этого электрические центры тяжести положительных зарядов ядер и отрицательных
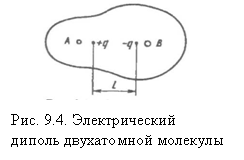
зарядов электронов не совпадают, и в молекуле возникает постоянный электрический диполь система двух равных по величине и противоположных по знаку зарядов +q и - q, разделенных расстоянием ℓ, называемым длиной диполя (рис. 8.4). Взаимодействие молекулы с внешним электрическим полем будет зависеть от величины вектора μ — электрического дипольного момента молекулы
μ = qℓ (9.1)
В квантовой механике дипольным моментом молекулы называется величина
![]()
![]() ,
,
 |
где ρ(r) — плотность заряда молекулы.
Вектор μ, так же как и ℓ, направлен от положительного к отрицательному полюсу.
Дипольный момент — наиболее непосредственная характеристика полярности связи. Неполярны (μ= 0) двухатомные гомонуклеарные молекулы (чисто ковалентная связь), в гетеронуклеарных молекулах связь полярна (μ ≠ 0). Особенно велики значения μ у ионных молекул. Неполярны многоатомные молекулы второго периода, молекулы, имеющие центр симметрии (BeF2, SF6 и др ) или обладающие высокой симметрией, например тетраэдрические — СН4, ССl4, плоские треугольные —BF3. Измерение дипольного момента может дать представление о симметрии молекулы.
Так, полярность молекулы Н2О указывает на ее изогнутость, а неполярность молекулы СO2 — на линейность. Дипольный момент многоатомной молекулы можно условно представить как векторную сумму дипольных моментов, приписываемых отдельным связям (аддитивность дипольного момента) Так, ![]() можно представить как векторную сумму дипольных моментов двух связей О—Н:
можно представить как векторную сумму дипольных моментов двух связей О—Н:
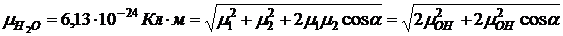 ,
,
Откуда 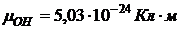
Поляризация. Если неполярную молекулу, атом или ион поместить в электрическое поле, то электрические центры тяжести положительных и отрицательных зарядов, совпадавшие до этого, смещаются относительно прежнего положения и отстоят теперь на расстоянии ℓ друг от друга. Это явление называют поляризацией. В частице возникает индуцированный (наведенный) дипольный момент
μинд = qℓ (9.2)
Зависимость ![]() от напряженности поля E можно представить в виде степенного ряда
от напряженности поля E можно представить в виде степенного ряда
![]() (9.3)
(9.3)
При малых полях пренебрегают всеми членами (8.3), кроме первого, тогда величина
![]() (9.4)
(9.4)
Коэффициент пропорциональности α в (8.4) называют поляризуемостью молекул. Поляризуемость в системе СГС имеет размерность объема
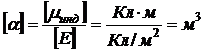 .
.
В системе СИ та же размерность для α следует из определения
![]() ,
,
где ![]() — электрическая постоянная. Величина поляризуемости молекул имеет порядок l Å3(lÅ3 = 10-30 м3) и характеризует объем электронного облака молекулы.
— электрическая постоянная. Величина поляризуемости молекул имеет порядок l Å3(lÅ3 = 10-30 м3) и характеризует объем электронного облака молекулы.
Поляризуемость, связанная с деформацией частицы, называется деформационной. Она характеризует смещение электронного облака и ядер относительно прежних положений, а также смещение атомных групп, таких, как NO2, ОН и т. п., и поэтому представляет собой сумму электронной и атомной поляризуемости: 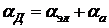 .
.
Так как ядра менее подвижны, чем электроны, то атомной поляризуемостью часто пренебрегают и принимают 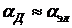 . Считая молекулу проводящей сферой радиуса r, можно показать, что электронная поляризуемость молекулы равна
. Считая молекулу проводящей сферой радиуса r, можно показать, что электронная поляризуемость молекулы равна 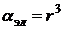 , таким образом, объем молекулы -
, таким образом, объем молекулы - ![]() . Чем больше электронов в молекуле, тем больше ее объем, тем больше и поляризуемость. Электронная поляризуемость экспериментально определяется через молярную рефракцию Rm:
. Чем больше электронов в молекуле, тем больше ее объем, тем больше и поляризуемость. Электронная поляризуемость экспериментально определяется через молярную рефракцию Rm:
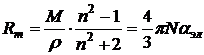 . (9.5)
. (9.5)
Измерив показатель преломления вещества n, и зная его плотность ρ и молекулярную массу М, можно по (8.5) рассчитать рефракцию Rm и поляризуемость ![]() .
.
Деформационная поляризация характерна для всех молекул. Полярные молекулы помимо деформационной поляризации испытывают во внешнем поле еще и ориентационную поляризацию, т. е. стремятся ориентировать свой постоянный диполь в направлении силовых линий поля. Этот эффект характеризуется ориентационной поляризуемостью ![]() , обратно пропорциональной абсолютной температуре:
, обратно пропорциональной абсолютной температуре:
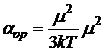 . (9.6)
. (9.6)
Как следует из (8.6), тепловое движение препятствует ориентации молекул в поле. Полная поляризуемость полярных молекул
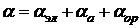 , (9.7)
, (9.7)
связана с диэлектрической проницаемостью ![]() вещества соотношением, которое называется уравнением Клаузиуса – Мосотти:
вещества соотношением, которое называется уравнением Клаузиуса – Мосотти:
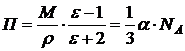 (9.8)
(9.8)
функция П, называемая молярной поляризацией, зависит от температуры [см. (]: П = А + В/Т, где ![]() ,
, 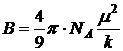 .
.
Измеряя температурную зависимость диэлектрической проницаемости газа, можно найти дипольный момент его молекул и поляризуемость. От поляризуемости молекул зависит так называемое дисперсионное взаимодействие атомов и молекул, которое играет важную роль в свойствах жидкостей и растворов, в процессах адсорбции, конденсации и др. Взаимодействие молекул с электромагнитным полем тем больше, чем больше их поляризуемость.
Связанная с поляризуемостью рефракция используется в структурной химии. Рефракция молекулы может быть представлена как сумма рефракций составляющих ее атомов (аддитивность рефракции). При этом учитываются дополнительные слагаемые (инкременты) для двойной, тройной связи и т. д. Помимо систем атомных рефракций используются системы рефракций связей.
Сравнивая экспериментальную рефракцию с её значением, вычисленным по аддитивной схеме, можно сделать вывод о строении молекул данного вещества.
При наличии сопряженных связей в открытых цепях органических молекул наблюдается заметное превышение экспериментальной Rm над вычисленной (экзальтация рефракции).
Насыщаемость ковалентной связи
Одним из важных свойств химической (ковалентной) связи в молекулах с закрытыми оболочками является ее насыщаемость. Так, из атомов водорода может образоваться молекула Н2, но не Н3 или Н4. Причина насыщаемости химической связи заключается в самой природе атомов и молекул как многоэлектронных систем в подчинении к принципу Паули.
Если две молекулы Н2 в основном состоянии оказываются очень близко друг к другу, два электрона первой молекулы на ![]() -орбитали и два электрона второй молекулы на такой же
-орбитали и два электрона второй молекулы на такой же ![]() орбитали оказываются в одной области пространства, то МО перекрываются. При этом данному электрону первой молекулы отвечает во второй молекуле электрон в точно таком же квантовом состоянии. Такие два электрона, согласно принципу Паули, будут избегать друг друга, и обе пары электронов сблизившихся молекул будут стремиться уйти из области соприкосновения, уводя с собой ядра, т. е. будет наблюдаться отталкивание молекул. Слияние системы в молекулу Н4 не произойдет. Связь в молекулах H2 в этом смысле насыщена. Аналогичное состояние отталкивания, часто называемое обменным отталкиванием или отталкиванием Паули, возникает при сближении и других молекул.
орбитали оказываются в одной области пространства, то МО перекрываются. При этом данному электрону первой молекулы отвечает во второй молекуле электрон в точно таком же квантовом состоянии. Такие два электрона, согласно принципу Паули, будут избегать друг друга, и обе пары электронов сблизившихся молекул будут стремиться уйти из области соприкосновения, уводя с собой ядра, т. е. будет наблюдаться отталкивание молекул. Слияние системы в молекулу Н4 не произойдет. Связь в молекулах H2 в этом смысле насыщена. Аналогичное состояние отталкивания, часто называемое обменным отталкиванием или отталкиванием Паули, возникает при сближении и других молекул.
Однако понятие насыщаемости ковалентной связи нельзя рассматривать как абсолютное. Можно представить, что при сближении двух молекул Н2 внешнее воздействие преодолевает силы отталкивания и четыре электрона разместятся на двух новых орбиталях, охватывающих все четыре ядра водорода. Расчет (Конрой и Малли, 1969) показал, что энергия такой системы на 523 кДж/моль превышает энергию двух молекул Н2. Это значит, что орбитали системы Н4 лежат намного выше, чем у молекул Н2. Такая система, будучи предоставлена самой себе, окажется неустойчивой по отношению к распаду на две молекулы Н2.
Следовательно, насыщаемость — понятие относительное, оно связано с тем, насколько высоко лежат молекулярные орбитали, на которые должны переходить электроны сливающихся молекул. Если энергия этих орбиталей достаточно низка, то молекулы, которые принято считать валентно насыщенными, могут образовать устойчивые соединения с другими молекулами или атомами. К таким соединениям относятся соединения с так называемой донорно-акцепторной связью.
§ 3. Донорно-акцепторная связь
Для образования донорно-акцепторого соединения между двумя частицами с закрытыми оболочками одна из них (акцептор электронов) должна иметь низшую свободную орбиталь, другая (донор электронов) — внешнюю несвязывающую орбиталь, заполненную двумя электронами. Перекрывание этих двух орбиталей приводит к образованию двух новых МО, общих для всей системы, и возникновению нового химического соединения (рис. 8.5).
Образовавшаяся связь называется донорно-акцепторной связью. Она осуществляется за счет перехода электронов донора на общую МО соединения, их обобществления. При этом на доноре возникает некоторый избыточный положительный заряд (электроны смещаются с несвязывающей МО), а на акцепторе — отрицательный ![]() .
.
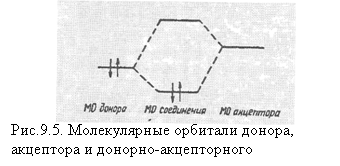
Таким образом, донорно-акцепторная связь по своей природе является ковалентной связью с той или иной степенью полярности. Название же "донорно-акцепторная связь" указывает лишь на механизм образования связи. Примером такой связи служит связь в комплексе BF3![]() NH3, где общая связывающая МО образуется за счет комбинации МО неподеленной пары молекулы NH3 (донор) и низколежащей свободной МО молекулы BF3 (акцептор). К донорно-акцепторным соединениям относятся соли тетрацианопарахинодиметана и тетратиофульвалена, так называемые "органические металлы", обладающие высокой металлической проводимостью.
NH3, где общая связывающая МО образуется за счет комбинации МО неподеленной пары молекулы NH3 (донор) и низколежащей свободной МО молекулы BF3 (акцептор). К донорно-акцепторным соединениям относятся соли тетрацианопарахинодиметана и тетратиофульвалена, так называемые "органические металлы", обладающие высокой металлической проводимостью.
Донорно-акцепторная связь возникает при образовании иона гидроксония Н3О+ между молекулой Н2О и ионом водорода, когда комбинирует свободная орбиталь (1s) водородного иона (акцептор) с МО неподеленной пары молекулы воды (донор). Все три водорода в Н3О+ совершенно равноценны, т. е. донорно-акцепторная связь в Н3О+ неотличима от ковалентной связи. Прочность донорно-акцепторной связи может быть велика: при образовании Н3О+ из Н+ и Н2O выделяется 710 кДж/моль, комплекс BF3NH3 перегоняется без разложения. Донорно-акцепторная связь может возникать и между атомами в кристаллах. Так, в кристалле InSb атом In предоставляет для связи вакантную низкую АО, а атом Sb — орбиталь неподеленной пары электронов.
Донорно-акцепторные соединения образуются и в растворах. Так, молекулярный йод, растворенный в СС14, образует D—А-комплексы с органическими соединениями — метанолом, диоксаном, пиридином и т. п. Здесь электроны неподеленной пары донора, например, пиридина, переходят на σ - связывающую орбиталь комплекса, а свободная ![]() разрыхляющая орбиталь молекулы I2 — акцептора — становится разрыхляющей орбиталью комплекса. Доказательством образования соединения служит появление полосы поглощения в ультрафиолетовой области спектра (2500 Å). Последняя вызвана переносом электрона σ-связывающей МО комплекса на
разрыхляющая орбиталь молекулы I2 — акцептора — становится разрыхляющей орбиталью комплекса. Доказательством образования соединения служит появление полосы поглощения в ультрафиолетовой области спектра (2500 Å). Последняя вызвана переносом электрона σ-связывающей МО комплекса на ![]() -разрыхляющую его орбиталь при поглощении кванта света. Подобные D-А соединения называют комплексами с переносом заряда.
-разрыхляющую его орбиталь при поглощении кванта света. Подобные D-А соединения называют комплексами с переносом заряда.
Лекция№ 10
Теория химической связи
План лекции:
1. Ионная связь. Степень полярности химической связи
2. Метод Хюккеля
3. Ионная связь в кристалле
§ 1. Ионная связь. Степень полярности химической связи
При очень высокой полярности связи (μ = 3·10-39Кл![]() м) электронный заряд на связывающей орбитали уже не распределен между двумя ядрами, а практически сосредоточен в области одного ядра, как, например, у ядра F в молекуле NaF. Здесь связывающая орбиталь мало отличается от атомной орбитали фтора
м) электронный заряд на связывающей орбитали уже не распределен между двумя ядрами, а практически сосредоточен в области одного ядра, как, например, у ядра F в молекуле NaF. Здесь связывающая орбиталь мало отличается от атомной орбитали фтора ![]() , т. е.
, т. е. ![]() . Волновая функция, приближенно описывающая два связующих электрона молекулы
. Волновая функция, приближенно описывающая два связующих электрона молекулы ![]() , указывает, что оба они движутся в поле ядра фтора.
, указывает, что оба они движутся в поле ядра фтора.
В результате вокруг ядра фтора сосредотачивается избыточный электрический заряд, практически равный единице; в то же время в силу электронейтральности молекулы ядро натрия оказывается центром равного по величине положительного заряда. Приближенно это положение может быть описано электростатической теорией ионной связи как перенос электрона от атома натрия к атому фтора с образованием ионов Na+ и F-, удерживаемых в молекуле электростатическими силами притяжения. В этом смысле предельное состояние связи при очень высокой полярности может быть названо ионной связью. Такого рода связь возникает в молекулах галогенидов щелочных металлов.
На основе представления об ионах в молекуле можно построить модель для расчета её свойств. Наиболее простая модель сферических ионов исходит из следующего. При сближении атомов Me и X происходит переход электрона от Me к X с образованием сферически симметричных ионов Me+ и X - (с внешней оболочкой s2р6). Такой переход всегда требует затраты энергии, равной ЭИ(Ме) — СЭ(Х). Последняя компенсируется энергией электростатического притяжения ионов, значительно превышающей затрату на ионизацию атомов, и это обеспечивает стабильность молекулы. Электростатические силы притяжения между ионами Me+ и X- не приводят к их слиянию, как было бы, будь ионы точечными зарядами. От слияния их удерживает отталкивание закрытых оболочек ионов. Энергию взаимодействия однозарядных ионов рассчитывают по формуле
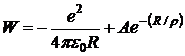 , (10.1)
, (10.1)
где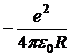 —энергия электростатического притяжения ионов;
—энергия электростатического притяжения ионов;
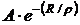 - энергия отталкивания закрытых оболочек; А и ρ - константы, определяемые на основе опыта. Баланс сил притяжения и отталкивания определяет равновесное расстояние между центрами ионов в молекуле R = rе, которому отвечает минимальное значение ее энергии We. Для вычисления We надо знать R = rе, А и ρ. Величины re молекул МеХ измерены с высокой точностью. Из условия минимума функции W при R=re
- энергия отталкивания закрытых оболочек; А и ρ - константы, определяемые на основе опыта. Баланс сил притяжения и отталкивания определяет равновесное расстояние между центрами ионов в молекуле R = rе, которому отвечает минимальное значение ее энергии We. Для вычисления We надо знать R = rе, А и ρ. Величины re молекул МеХ измерены с высокой точностью. Из условия минимума функции W при R=re
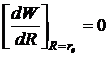 (10.2)
(10.2)
исключаем А из (9.1) и получаем
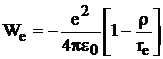 . (10.3)
. (10.3)
Достаточное условие минимума, 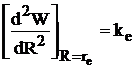 , где kе — константа квазиупругой силы молекулы, определяемая из спектров; отсюда находим ρ = 0,34
, где kе — константа квазиупругой силы молекулы, определяемая из спектров; отсюда находим ρ = 0,34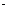 10-10 м (средняя величина у молекул Ме+Х-). Окончательно
10-10 м (средняя величина у молекул Ме+Х-). Окончательно
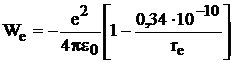 . (10.4)
. (10.4)
Энергия диссоциации молекулы на ионы равна энергии взаимодействия ионов с обратным знаком:.![]() .
.
Энергию диссоциации молекулы на нейтральные атомы De можно вычислить исходя из термохимического цикла:
![]() . (10.5)
. (10.5)
В качестве примера рассмотрим расчет De для молекулы NaCl с rе = 2,3606![]() м. Зная, что kе = 1,Н/м, найдем р = 0,29
м. Зная, что kе = 1,Н/м, найдем р = 0,29![]() м и по (9.4) We = -8,58
м и по (9.4) We = -8,58![]() Дж. Отсюда
Дж. Отсюда 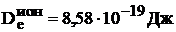 = 5,36 эВ.
= 5,36 эВ.
Величина 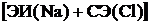 = 1,38 эВ. Энергия диссоциации согласно (10.5)
= 1,38 эВ. Энергия диссоциации согласно (10.5)
De = 5,36 - 1,38 = 3,98 эВ.
Опыт дает De(NaCl) = 4,23 эВ. Согласие надо считать удовлетворительным для такой грубой модели, как модель сферических ионов.
Величина 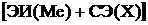 положительна для всех без исключения галогенидов щелочных металлов и согласно (9.13)
положительна для всех без исключения галогенидов щелочных металлов и согласно (9.13) 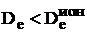 . Поэтому ионные молекулы в основном состоянии всегда диссоциируют на атомы, а не на ионы.
. Поэтому ионные молекулы в основном состоянии всегда диссоциируют на атомы, а не на ионы.
Рассмотренная модель сферических ионов позволяет выполнять расчеты и других молекулярных параметров помимо энергии диссоциации, например дипольного момента. Небольшие расхождения с опытом объясняются несовершенством модели. Квантовомеханические расчеты показывают, что хотя распределение электронной плотности в молекулах МеХ напоминает распределение для двух сферических ионов, однако автономных оболочек двух ионов в молекуле нет, как нет и полного переноса электрона от Me к X.
На основании таких расчетов построены диаграммы разности электронной плотности 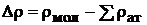 .
.
Для двух молекул с чисто ковалентной (N2) и ионной (LiF) связью имеем следующий результат. У молекулы N2 в области между ядрами наблюдается увеличение электронной плотности в сравнении с атомным распределением. Её симметричное распределение относительно центра молекулы — характерная черта чисто ковалентной связи в гомонуклеарных молекулах. В молекуле LiF область повышенной электронной плотности распределена резко асимметрично и сосредоточена почти целиком вокруг ядра фтора. Произошел почти полный перенос заряда одного электрона от атома металла к атому галогена. Эта особенность характерна для ионной связи. В молекулах с полярной ковалентной связью (например, HF) распределение электронной плотности — промежуточное между двумя предельными распределениями: чисто ковалентной и ионной связями.
Полярность связи и концепция электроотрицательности. Степень полярности связи наиболее непосредственно характеризуется дипольным моментом, часто для этого используется также концепция электроотрицательности. Полинг назвал электроотрицательностью атома (ЭО) способность его в молекуле притягивать на себя электрон Полярность молекулы определяется разностью электроотрицательностей атомов (ΔЭО), чем она выше, тем полярнее связь.
ЭО атома тем выше, чем выше его полярная связь (способность удержать свой электрон) и чем выше СЭ (способность притягивать электрон соседнего атома).
Поэтому мерой электроотрицательности может служить сумма ПИ+СЭ.
§2. Метод Хюккеля
При описании электронной структуры многоатомных молекул используют различные расчетные методы приближения МО ЛКАО.
Наиболее простым из них является полуэмпирический метод Хюккеля. В данном приближении коэффициенты ci многоцентровой МО, охватывающей k ядер 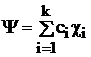 определяются по вариационному принципу. Для этого записывают вековой определитель
определяются по вариационному принципу. Для этого записывают вековой определитель
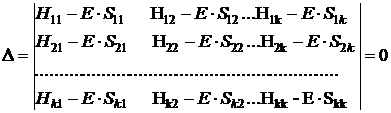 .
.
Он имеет k корней. В результате решения получается k МО (Ψ1, Ψ2,… Ψk), которым соответствует спектр собственных значений энергии E1, E2,…Ek.
Ввиду математической сложности вычисления соответствующих интегралов в методе Хюккеля вводятся следующие ограничения:
1) Все диагональные матричные элементы 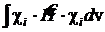 равны между собой, т. е. H11=H22=….Hkk=α. При этом постулируется, что кулоновский интеграл α равен энергии электрона в свободном атоме.
равны между собой, т. е. H11=H22=….Hkk=α. При этом постулируется, что кулоновский интеграл α равен энергии электрона в свободном атоме.
2) Все недиагональные элементы (обменные интегралы) ![]() равны нулю, если χi и χj являются АО несоседних атомов, т. е. (Hij(несосед)=0).
равны нулю, если χi и χj являются АО несоседних атомов, т. е. (Hij(несосед)=0).
3) Обменные интегралы всех соседних атомов постоянны и равны (Hij(сосед)=β).
4) Интегралы перекрывания Sij=0, если i≠j.
5) Для нормированных АО Sii=0.
6) В основном состоянии 2n электронов молекулы занимают n наиболее низких атомных орбиталей.
В этом постулате предполагается, что в форме орбитального приближения
![]()
При расчёте полной электронной энергии молекулы последние два члена компенсируют друг друга.
В этом же приближении электронная энергия атомов равна сумме орбитальных энергий, или E=2nα.
Энергия диссоциации равна De=-mβ, где β –эмпирический коэффициент.
7) Кулоновский и обменный интегралы α и β аналитически не вычисляются. Они рассматриваются как параметры. Для их оценки сравнивают результаты расчёта энергии в единицах β и экспериментальные данные. Частью интегралов полностью пренебрегают.
Таким образом, метод Хюккеля является полуэмпирическим. В данном методе при объяснении свойств молекул и химической активности её отдельных участков используют понятие электронной плотности заряда на атомах ξ, порядка связи ρ и индекса свободной валентности F.
Все эти характеристики рассчитывают через коэффициенты ci молекулярных орбиталей. Так как метод расчёта МО Хюккеля является приближённым, то эти величины весьма условны.
§3 Ионная связь в кристалле
Важнейшей особенностью ионной связи является ее ненасыщаемость и ненаправленность. Поле, создаваемое ионом, имеет сферическую симметрию, и все находящиеся в этом поле другие ионы испытывают его действие. В результате ионы в кристалле образуют трехмерную беспредельную решетку, в узлах которой правильно чередуются катионы и анионы. Отдельных молекул в решетках солей типа КСℓ нет.
Кристаллическую решетку ионного соединения можно рассматривать как бесконечное повторение минимального трехмерного участка (параллелепипеда), называемого элементарной ячейкой. В сответствии с симметрией элементарной ячейки кристаллическую решетку относят к одной из кристаллических систем (сингоний): кубической, тетрагональной, гексагональной, тригональной, орторомбической, моноклинной и триклинной (в порядке убывания симметрии). Ненасыщаемость и ненаправленность ионной связи приводят в большинстве ионных кристаллов к образованию структур так называемых плотнейших упаковок. Это кубические решетки типов NaСℓ и CsCℓ (рис. 4.25), фалерита (ZnS) и флюорита (СаРг), гексагональные типа ZnO и др.
Важной характеристикой структурного типа кристалла является координационное число (КЧ); для ионных кристаллов это число ионов противоположного знака, окружающих данный ион: для KCL/
В настоящее время надежно установлены теплоты образования и термодинамические функции многих кристаллов, а также ионов в газовой фазе, и термодинамические расчеты проводят, не прибегая к формуле (4.19). Но свой познавательный интерес она сохранила.
Лекция №11
Спектральные методы исследования строения и энергетических состояний молекулы
План лекции:
1. Ангармонизм колебательного спектра. Структура колебательного спектра
2. Электронные спектры. Определение энергии диссоциации двухатомных молекул.
3. Вращательные спектры и строение многоатомных молекул.
§1. Ангармонизм колебательного спектра. Структура колебательного спектра
Модель гармонического осциллятора полезна для понимания основных особенностей колебаний молекул. Но она очень неточна по следующим причинам. Потенциальная кривая гармонического осциллятора является бесконечной параболой, т. е. Eкол →∞.
Потенциальная кривая реальной молекулы имеет вид
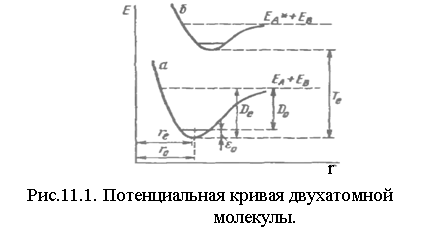
Dо - опытная мера прочности химической связи. Do – энергия диссоциации, отсчитываемая от дна потенциальной кривой.
![]() .
.
В двухатомной молекуле, если ей сообщить энергию E≥Do, то она разрывается на два невзаимодействующих атома. В отличие от эквидистантных линий гармонического осциллятора в реальной молекуле с увеличением колебательного квантового числа υ

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 |
