
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Так как А — функция состояния, то согласно теореме Коши значение ее производных не зависит от порядка дифференцирования. Отсюда, приравнивая вторые производные, получаем уравнение
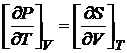 , (16.41)
, (16.41)
которое позволяет рассчитать увеличение энтропии при изотермическом расширении системы:
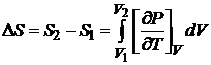 . (16.42)
. (16.42)
Первая производная 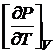 может быть определена из уравнения состояния.
может быть определена из уравнения состояния.
Если учесть (16.40), то выражение для энергии Гельмгольца можно записать также в виде
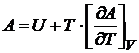 . (16.43)
. (16.43)
Уравнение (16.43) называется уравнением Гиббса — Гельмгольца. Для квазистатического процесса при постоянном объеме и температуре уравнение Гиббса - Гельмгольца примет вид
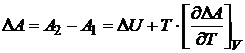 . (16.44)
. (16.44)
Так как максимальная работа для квазистатического процесса при рассматриваемых условиях (W'квази) =-(ΔА)V, T то
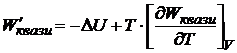 . (16.45)
. (16.45)
Свободная энергия широко используется в термодинамике, когда в качестве независимых переменных выбраны V и Т, которые легко определяются из эксперимента. Рассмотрим самопроизвольный процесс при Р = const и T= const. Из уравнения (16.27) следует ![]() . Введем обозначение
. Введем обозначение
G = U - TS + PV = A + PV = Н - TS. (16.46)
G является функцией состояния системы и называется энергией Гиббса. Энергия Гиббса при постоянных Р и Т является экстенсивным свойством системы. Для квазистатического процесса
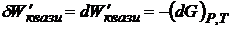 . и
. и 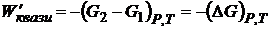 . (16.47)
. (16.47)
Отсюда для квазистатического процесса при рассматриваемых условиях максимальная (полезная) работа (W΄квази) приобретает свойства функции состояния и равна убыли энергии Гиббса. Для нестатического процесса
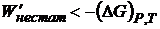 (16.48)
(16.48)
Продифференцируем выражение G = А + PV:
dG = dA+ PdV+ VdP.
Подставляя dA из (16.39), получим
dG = VdP-SdT. (16.49)
Из уравнения (16.49) следует, что энергия Гиббса есть явная и характеристическая функция переменных Р и Т. Так как dG — полный дифференциал, то
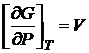 и
и 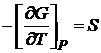 . (16.50)
. (16.50)
Из соотношений (8.50) следует, что объем системы служит мерой возрастания энергии Гиббса с возрастанием давления при постоянной температуре, а энтропия — мерой убыли энергии Гиббса с возрастанием температуры при постоянном давлении.
Из свойств полного дифференциала dG также вытекает
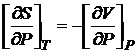 . (16.51)
. (16.51)
Из (16.51) следует
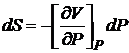 ,
,
а для конечного изменения энтропии при изменении давления от P1 до P2
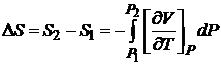 . (16.53)
. (16.53)
Уравнение (16.53) позволяет рассчитать изменение энтропии при изотермическом сжатии (Р2 > Р1) или расширении (Р2 < Р1) газа; первая производная 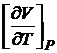 может быть определена из уравнения состояния.
может быть определена из уравнения состояния.
Подставляя S выражение из (16.50) в уравнение G = H – TS, получаем
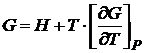 . (16.54)
. (16.54)
Для квазистатического процесса в закрытой системе при постоянном давлении и температуре
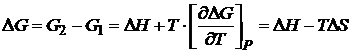 , (16,55)
, (16,55)
где ΔH-— изменение энтальпии; ΔS —- изменение энтропии. Так как максимальная полезная работа для квазистатического процесса при рассматриваемых условиях (W'квази) =-(ΔG)P, T, то
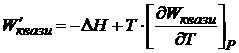 . (16.56)
. (16.56)
Уравнения (16.54) — (16.56) также называются уравнениями Гиббса - Гельмгольца. Энергия Гиббса широко используется в термодинамике, если в качестве независимых параметров выбраны Р и Т.
Параметры Р и T, как V и Т, легко определяются экспериментальным путем. Если химическая реакция протекает при постоянном давлении и температуре термодинамически необратимо, то ΔH равно тепловому эффекту Q. Следовательно, величину ΔН в уравнении (16.55) можно определить термохимическим способом (калориметрически или вычислить на основании закона Гесса).
Произведение T∙ΔS согласно уравнению (16.8) равно количеству теплоты, полученному или отданному системой. Таким образом, система должна находиться в тепловом контакте с внешней средой, температура которой постоянна и равна температуре в системе. Энтропию согласно уравнению (16.30)
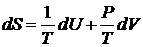
Также можно рассматривать как характеристическую функцию S = f(U, V). Итак, в зависимости от условий протекания квазистатического процесса максимальная полезная работа равна убыли термодинамических функций U, G, Н, и А:
![]() ;
; 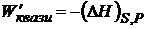 ;
; 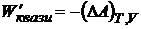 ;
; 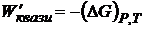 .
.
По аналогии с классической с механикой, где работа равна уменьшению потенциальной энергии системы, характеристические функции U = f(S, V); H = f(S, P); А = f(V, T) и G = f(P, T) также называются термодинамическими потенциалами.
Исходя из рассмотренных свойств характеристических функций U, H, A, G, внутреннюю энергию можно назвать изохорно - изоэнтропийным потенциалом, энтальпию - изобарно – изоэнтропийным потенциалом, энергию Гельмгольца – изохорно - изотермическим и энергию Гиббса - изобарно – изотермическим потенциалом.
Лекция №16
Химическая термодинамика
План лекции:
1. Изменение энтропии идеального газа.
2. Энергия Гиббса смеси идеальных газов. Химический потенциал.
3. Общие условия химического равновесия.
§1. Изменение энтропии идеального газа.
а) Объединённое уравнение 1 – ого и 2 – го законов термодинамики для квазистатического процесса в одном моле идеального газа записывается в виде
T∙dS = CV∙dT + P∙dV = CV∙dT + R∙T∙dV/V, или
T∙dS = CP∙dT - V∙dP = CP∙dT - R∙T∙dP/P, откуда
![]() и
и ![]() .
.
Считая CV и СP независящими от температуры после интегрирования получим уравнения
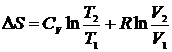 и
и 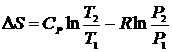 , которые позволяют рассчитывать изменение энтропии одного моля идеального газа при переменных T и V, и при переменных T и P. В частности, при изотермическом расширении идеального газа
, которые позволяют рассчитывать изменение энтропии одного моля идеального газа при переменных T и V, и при переменных T и P. В частности, при изотермическом расширении идеального газа  , при изохорическом нагревании
, при изохорическом нагревании 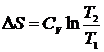 , а при изобарическом процессе
, а при изобарическом процессе 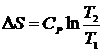 .
.
Помимо вычисления изменения энтропии интерес представляет и значение самой величины S для одного моля идеального газа. Положим P1 = P0 и T1 = 1K.
Тогда 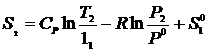 , где
, где ![]() - стандартная энтропия при 1K, которая называется энтропийной постоянной идеального газа. Отбросим индекс 2,а
- стандартная энтропия при 1K, которая называется энтропийной постоянной идеального газа. Отбросим индекс 2,а ![]() запишем в виде S,́ тогда
запишем в виде S,́ тогда
![]() , (17.1)
, (17.1)
где ![]() - относительное давление. Стоящие под знаком логарифма величины T = T/1 и
- относительное давление. Стоящие под знаком логарифма величины T = T/1 и 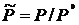 являются безразмерными величинами. Энтропийную постоянную Ś можно вычислить на основании законов статистической термодинамики по молекулярным постоянным. Зная величину
являются безразмерными величинами. Энтропийную постоянную Ś можно вычислить на основании законов статистической термодинамики по молекулярным постоянным. Зная величину ![]() , по формуле (17.1) можно вычислить абсолютные значения энтропий для одного моля идеального газа.
, по формуле (17.1) можно вычислить абсолютные значения энтропий для одного моля идеального газа.
б) Вычислим изменение энтропии при смешении химически невзаимодействующих идеальных газов при нестатическом процессе.
Представим себе цилиндр, разделённый перегородками на k ячеек. Объём цилиндра V равен сумме объёмов ячеек 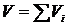 , где Vi – объём i–й ячейки. Допустим, что в i - й ячейке находится νi молей идеального газа. i-го типа. Будем считать, что давление P и температура T газа в каждой ячейке одинаковы. В соответствии с (17.1) энтропия i-й компоненты газа равна
, где Vi – объём i–й ячейки. Допустим, что в i - й ячейке находится νi молей идеального газа. i-го типа. Будем считать, что давление P и температура T газа в каждой ячейке одинаковы. В соответствии с (17.1) энтропия i-й компоненты газа равна
![]() . (17.2)
. (17.2)
Так как энтропия является экстенсивным свойством системы, то для νi молей в ячейке i она будет равна
![]() , а энтропия всей исходной системы будет равна сумме энтропий отдельных газов
, а энтропия всей исходной системы будет равна сумме энтропий отдельных газов
![]() . (17.3)
. (17.3)
Если разрушить перегородки, то компоненты газа вследствие протекания нестатического процесса диффузии через некоторое время смешаются, и каждый газ займёт весь объём цилиндра. Так как газы являются идеальными, то температура не изменится. Согласно закону Дальтона 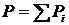 .
.
С другой стороны для идеальной газовой смеси имеем: ![]() ,
, 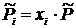 , где xi – молярная доля i – ого компонента газовой смеси. В идеальной смеси каждый газ смеси ведёт себя независимо от других газов. Тогда в соответствии с уравнением (17.1) энтропия моля i - й компоненты газовой смеси равна
, где xi – молярная доля i – ого компонента газовой смеси. В идеальной смеси каждый газ смеси ведёт себя независимо от других газов. Тогда в соответствии с уравнением (17.1) энтропия моля i - й компоненты газовой смеси равна ![]() , а энтропия системы в конечном состоянии равна
, а энтропия системы в конечном состоянии равна 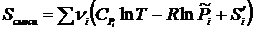 .
.
Так как энтропия является функцией состояния, то её изменение в результате смешивания газов равно разности  . Или, в явном виде
. Или, в явном виде 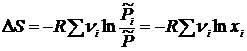 . Отсюда, изменение энтропии при образовании 1 моля смеси идеальных газов будет равно
. Отсюда, изменение энтропии при образовании 1 моля смеси идеальных газов будет равно
![]() .
.
Согласно последнему уравнению величина ΔS>0, так как xi<1 и lnxi < 0. Следовательно, при смешении идеальных газов энтропия системы будет возрастать.
§2. Энергия Гиббса смеси идеальных газов. Химический потенциал
Для вычисления энергии Гиббса смеси идеальных газов необходимо установить взаимосвязь между внутренней энергией U, произведением P∙V, энтропией S и составом системы.
Предположим, что равновесная газовая смесь содержит k различных веществ, и все они находятся в идеальном газовом состоянии. В смеси идеальных газов, как внутренняя энергия, так и энтропия системы являются аддитивными функциями состава. Рассмотрим вначале первое слагаемое в выражении для энергии Гиббса. Бесконечно малое изменение 1 моля i - го компонента равно
![]() , (17.4)
, (17.4)
где ![]() - молярная теплоёмкость i–й компоненты смеси. Будем считать в первом приближении, что
- молярная теплоёмкость i–й компоненты смеси. Будем считать в первом приближении, что ![]() не зависит от температуры. Интегрируя (17.1) получим
не зависит от температуры. Интегрируя (17.1) получим
![]() (17.5)
(17.5)
где ![]() - внутренняя энергия 1 моля i-й компоненты при 0K. Если газовая смесь содержит νi моль i – го газа, то умножая обе части уравнения (17.5) на νi и суммируя по всем компонентам смеси, будем иметь
- внутренняя энергия 1 моля i-й компоненты при 0K. Если газовая смесь содержит νi моль i – го газа, то умножая обе части уравнения (17.5) на νi и суммируя по всем компонентам смеси, будем иметь
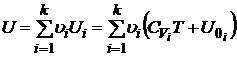 . (17.6)
. (17.6)
Используя уравнение Менделеева – Клапейрона запишем второе слагаемое энергии Гиббса
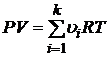 . (17.7)
. (17.7)
Рассмотрим третье слагаемое. В соответствии (17.2) зависимость энтропии Si одного моля i – й компоненты газовой смеси от относительного парциального давления ![]() и температуры
и температуры
![]() .
.
Умножая обе части последнего уравнения на νi и суммируя по всем k компонентам, получим
![]() .
.
Подставив значения U, PV, и S в правую часть выражения для энергии Гиббса, имеем
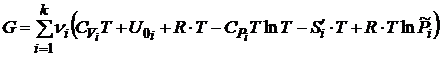 . (17.8)
. (17.8)
Первые пять слагаемых в уравнении (17.8) зависят от природы индивидуального i – го вещества и температуры и не зависят от состава смеси и давления. Алгебраическую сумму этих пяти слагаемых обозначим через ![]() . Тогда
. Тогда
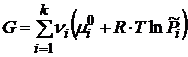 , (17.9)
, (17.9)
или
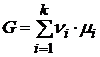 . (17.10)
. (17.10)
Величина
![]() (17.11)
(17.11)
Называется химическим потенциалом индивидуального i – ого вещества в газовой смеси, а величина ![]() - стандартным потенциалом (при
- стандартным потенциалом (при ![]() ). Так как для идеальной газовой смеси
). Так как для идеальной газовой смеси 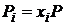 и
и 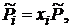 то уравнение (17.11) можно привести к виду
то уравнение (17.11) можно привести к виду
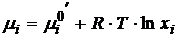 (17.12)
(17.12)
Если ![]() является функцией только температуры, то
является функцией только температуры, то ![]() является функцией температуры и давления. Подставив значения в выражение (17.8 ) получим
является функцией температуры и давления. Подставив значения в выражение (17.8 ) получим
![]() и
и ![]() . (17.13)
. (17.13)
Из последних уравнений следует, что химический потенциал равен энергии Гиббса, приходящейся на 1 моль i – ой компоненты газовой смеси.
Таким образом, химический потенциал это парциальная молярная энергия Гиббса.
При протекании химических процессов в газовой системе происходит изменение её состава. Если процесс протекает при постоянных P и T, то для системы с переменным составом для бесконечно малого изменения энергии Гиббса получим
![]() . (17.14)
. (17.14)
Второе слагаемое в (17.14) равно нулю, т. к. ![]() зависит только от температуры, то при T =const дифференциал
зависит только от температуры, то при T =const дифференциал ![]() равен 0. Рассмотрим второе слагаемое
равен 0. Рассмотрим второе слагаемое ![]() .
.
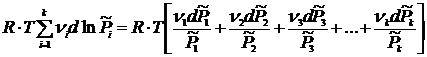 .
.
Учитывая, что 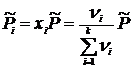 , получим
, получим
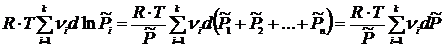 .
.
При постоянном давлении 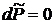 , таким образом, утверждение о равенстве 0 второго слагаемого в (17.14) доказано.
, таким образом, утверждение о равенстве 0 второго слагаемого в (17.14) доказано.
Таким образом, для системы переменного состава
![]() . (17.15)
. (17.15)
Полный дифференциал энергии Гиббса в соответствии с определением и с учётом (17. ) можно записать следующим образом
![]() . (17.16)
. (17.16)
Исходя из уравнения (17.16) и используя уравнение (16.46) можно показать справедливость следующих выражений для систем с переменным составом:
![]() , (17.17)
, (17.17)
![]() , (17.18)
, (17.18)
![]() . (17.19)
. (17.19)
В качестве примера докажем уравнение (17.17). Дифференцируя выражение G=A+PV, получим
dG=dA+PdV+VdP. (17.20)
Из сопоставления формул (17.18) и (17.19) находим ![]() . Откуда
. Откуда ![]() .
.
Из уравнений (17.16) и (17.20) следует
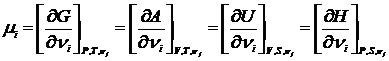 . (j≠i) (17.21)
. (j≠i) (17.21)
Таким образом, химический потенциал является частной производной по количеству i - ой компоненты газовой смеси при постоянном количестве остальных индивидуальных компонент и при постоянстве соответствующих независимых переменных.
Так как характеристические функции представляют экстенсивные свойства системы, то химические потенциалы, согласно уравнению (17.21) являются парциальными молярными величинами. Нужно отметить, что химический потенциал является интенсивным свойством системы [см. (17.10)].
Химические потенциалы широко применяются не только в химической термодинамике газовых смесей, но и при изучении растворов, фазовых равновесий, равновесных электродных потенциалов.
§3. Общие условия химического равновесия
Рассмотрим в общем виде газофазную реакцию
ν1A1 + ν2A2 = ν3A3 + ν4A4
при постоянном давлении и постоянной температуре. Допустим, что в момент приготовления реакционной смеси имеются исходные вещества и продукты реакции. Обозначим количества вещества в молях в системе в этот момент времени для реагентов A1, A2, A3 и A4 через ![]() ,
, ![]() ,
, ![]() и
и![]() , а в процессе реакции – соответственно через n1, n2, n3 и n4. При протекании реакции слева направо количества исходных веществ будет уменьшаться, а количества продуктов реакции увеличиваться. Изменение количеств реагирующих веществ в процессе реакции
, а в процессе реакции – соответственно через n1, n2, n3 и n4. При протекании реакции слева направо количества исходных веществ будет уменьшаться, а количества продуктов реакции увеличиваться. Изменение количеств реагирующих веществ в процессе реакции ![]() ,
, ![]() ,
, 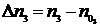 ,
, 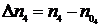 связаны между собой соотношением
связаны между собой соотношением
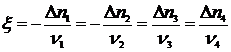 . (17.22)
. (17.22)
Отсюда
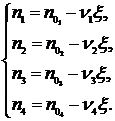 (17.23)
(17.23)
Соотношение 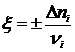 , (17.24)
, (17.24)
называется химической переменной. Она определяет число пробегов реакции. Химическая переменная равна нулю, если реакция ещё не протекала и равна 1, если ν1 и ν2 моль исходного вещества превратились в ν3 и ν4 моль продуктов реакции. Итак, в пределах одного пробега реакции химическая переменная может принимать значения от 0 до 1.
Например, если при синтезе аммиака 3H2 = N2 = 2NH2 за какой-то промежуток времени прореагирует 3 моль водорода и 1моль азота, то ξ = 1, если же прореагирует 1,5 моль водорода и 0,5 моль азота, то ξ = 0,5.
Дифференцируя соотношение (17.23), получим
dni = ±νi∙dξ. (17.24)
Подстановка выражения (14.24) для dni в уравнение (17.10) для квазистатического процесса приводит к соотношению
![]() . (17.25)
. (17.25)
При установлении химического равновесия в системе ![]() . Отсюда
. Отсюда
![]() . (17.26)
. (17.26)
Так как химическая реакция протекала, то dξ≠0. Тогда
![]() . (17.27)
. (17.27)
Лекция № 17
Химическая термодинамика
План лекции:
1. Закон действующих масс. Константа равновесия для газофазных реакций.
2. Уравнение изотермы реакции.
3. Интегральная форма зависимости энергии Гиббса и стандартной константы равновесия от температуры
§1. Закон действующих масс. Константа равновесия для газофазных реакций
Пусть между газообразными веществами A1, A2, A3 и A4 протекает химически обратимая реакция ν1A1 + ν2A2 ↔ ν3A3 + ν4A4. При определённых P, T и концентрации реагентов скорость прямой реакции значительно превышает скорость обратной, и процесс протекает самопроизвольно в прямом направлении.
При постоянной температуре и давлении скорости прямой и обратной реакций постоянно выравниваются в результате изменений концентраций реагирующих веществ. Химическая реакция протекает самопроизвольно до достижения химического равновесия между реагирующими веществами. Химическое равновесие является динамическим равновесием, в результате которого скорости прямой и обратной реакций равны друг другу.
Состояние химического равновесия характеризуется двумя признаками:
1) если система находится в состоянии равновесия, то её состав при постоянных условиях не изменяется с течением времени;
2) если система, находящаяся в равновесии будет выведена из этого состояния вследствие внешних воздействий, то после прекращения их действия она возвращается в исходное состояние.
При установлении химического равновесия имеет место соотношение
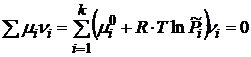 , (18.1)
, (18.1)
Отсюда получим
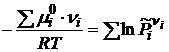 . (18.2)
. (18.2)
В выражении суммы, слагаемые для исходных веществ берутся со знаком «-», а для продуктов реакции со знаком «+». После потенцирования (18.2) получим
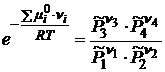 . (18.3)
. (18.3)
Так как ![]() зависит только от природы i-го индивидуального реагента и температуры, то при T=const левая часть в уравнении (18.3) является постоянной величиной и называется стандартной константой равновесия реакции
зависит только от природы i-го индивидуального реагента и температуры, то при T=const левая часть в уравнении (18.3) является постоянной величиной и называется стандартной константой равновесия реакции ![]() :
:
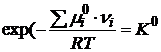 . (18.4)
. (18.4)
Стандартная константа равновесия является безразмерной величиной. Если составные части реакционной смеси подчиняются законам идеального газа, то в соответствии с (18.3)
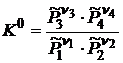 . (18.5)
. (18.5)
Уравнение (18.5) называется законом действующих масс. Для реальных систем, в которых нельзя пренебречь силами взаимодействия между молекулами, произведение равновесных парциальных давлений 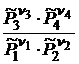 , обозначаемое далее как
, обозначаемое далее как 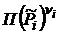 , будет изменяться с увеличением общего давления, т. е. уже не будет равно стандартной константе равновесия химической реакции.
, будет изменяться с увеличением общего давления, т. е. уже не будет равно стандартной константе равновесия химической реакции.
Таким образом, уравнение (18.5) можно использовать для расчёта химического равновесия в реальных газообразных системах только при невысоких давлениях. Если исходить из уравнения
![]() (18.6)
(18.6)
то таким же образом, как и при выводе (18.6) можно записать закон действующих масс в виде
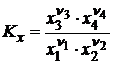 , (18.7)
, (18.7)
где Kx – константа равновесия, выраженная через молярные доли в момент равновесия. Так как ![]() зависит от температуры и давления, то Kx =f(T, P). Термодинамический вывод закона действующих масс был сделан Вант-Гоффом в 1885 г.
зависит от температуры и давления, то Kx =f(T, P). Термодинамический вывод закона действующих масс был сделан Вант-Гоффом в 1885 г.
Для газофазных реакций закон действующих масс можно записать ещё следующим образом
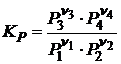 (18.8)
(18.8)
и 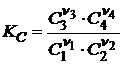 , (18.9)
, (18.9)
где Pi – парциальное давление; Ci – концентрация i - ого реагента в момент равновесия, измеренный в моль/л.
Константы KP и KC являются эмпирическими, поскольку они определяются через определяемые на опыте значения Pi и Ci.
Если константы равновесия K0 и Kx являются безразмерными величинами, то KP и KC величины размерные.
§2. Уравнение изотермы реакции
Предположим, что в смеси идеальных газов квазистатически протекает реакция ν1A1 + ν2A2 ↔ ν3A3 + ν4A4. Допустим, что в момент приготовления реакционной смеси имеются исходные вещества и продукты реакции. Обозначим относительные парциальные давления реагентов в системе в момент приготовления реакционной смеси через ![]() ,
, ![]() ,
,![]() и
и ![]() .
.
Если реакция совершила один пробег, т. е. ξ = 1, при постоянных P и T, то тепловой эффект реакции можно рассчитать по формуле
![]() . (18.10)
. (18.10)
Уравнение (18.10) можно преобразовать к виду
![]() . (18.11)
. (18.11)
Так как ![]() , то
, то
![]() . (18.12)
. (18.12)
Выражение (18.12) называется уравнением изотермы реакции. Величину ![]() называют мерой химического сродства.
называют мерой химического сродства.
Уравнение (18.12) позволяет вычислять изменение энергии Гиббса, а также предсказывать направление протекания реакции при заданных условиях, если известны стандартная константа равновесия и относительные парциальные давления реагирующих веществ в момент их смешивания. Константа равновесия K0 может быть вычислена на основании равновесного состава реакционной смеси или по третьему закону термодинамики.
При 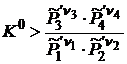 величина
величина ![]() . В этом случае самопроизвольное протекание реакции возможно только слева направо.
. В этом случае самопроизвольное протекание реакции возможно только слева направо.
Если же 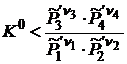 то
то ![]() и самопроизвольное протекание реакции будет возможно в направлении образования исходных веществ, то есть справа налево.
и самопроизвольное протекание реакции будет возможно в направлении образования исходных веществ, то есть справа налево.
Если оба слагаемых в правой части уравнения (18.12) равны между собой, то ![]() , что соответствует состоянию химического равновесия в закрытой системе.
, что соответствует состоянию химического равновесия в закрытой системе.
Следовательно, чтобы ответить на вопрос о возможности протекания химической реакции в закрытой системе, слева направо или справа налево, необходимо знать состав реакционной смеси в момент её приготовления и стандартную константу равновесия при данной температуре.
Если в момент смешивания относительные парциальные давления каждого реагента равны единице, т. е. все реагенты вступают в реакцию в своих стандартных состояниях, то уравнение (18.12) примет вид
![]() . (18.13)
. (18.13)
Уравнение (18.13) называют уравнением стандартного сродства.
Так как
![]() , (18.14)
, (18.14)
то уравнение (18.13) можно записать в виде
![]() . (18.15)
. (18.15)
Уравнения (18.14) и (18.15) широко используются при рассмотрении ряда вопросов химического равновесия.
Если при термодинамических расчётах используется эмпирическая константа равновесия KP, то мера химического сродства рассчитывается по формуле
![]() , (18.16)
, (18.16)
где P0 – стандартное давление.
§3. Интегральная форма зависимости энергии Гиббса и стандартной константы равновесия от температуры
Допустим, что между газообразными веществами A1, A2, A3 и A4 протекает химически обратимая реакция ν1A1 + ν2A2 ↔ ν3A3 + ν4A4.
Согласно закону действующих масс , где ![]() - относительное парциальное давление i-ой компоненты системы в состоянии химического равновесия.
- относительное парциальное давление i-ой компоненты системы в состоянии химического равновесия.
Для определения ![]() запишем уравнение Гиббса-Гельмгольца для стандартных условий
запишем уравнение Гиббса-Гельмгольца для стандартных условий
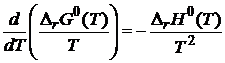 . (18.17)
. (18.17)
Комбинируя (18.13) и (18.17), получим
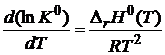 , (18.18)
, (18.18)
или
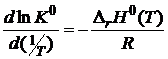 . (18.19)
. (18.19)
Уравнения (18.18) и (18.19) называются уравнениями Вант-Гоффа или уравнениями изобары реакции.
Лекция №18
Основы химической термодинамики
План лекции:
1. Тепловая теорема Нернста
2. Третий закон термодинамики
3. Расчёт изменения стандартной энергии Гиббса и константы равновесия по методу Тёмкина-Шварцмана
4. Расчёт изменения стандартной энергии Гиббса и константы равновесия с помощью функций приведённой энергии Гиббса
§1. Тепловая теорема Нернста
Зависимость изменения стандартной энергии Гиббса ![]() и стандартного теплового эффекта реакции
и стандартного теплового эффекта реакции ![]() от температуры может быть получена из уравнения Ван-Гоффта следующим образом
от температуры может быть получена из уравнения Ван-Гоффта следующим образом
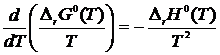
Интегрируя уравнение Вант-Гоффта по температуре, имеем
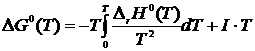 ,
,
Так как 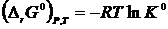 ,
,
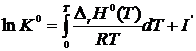 .
.
Из закона Кирхгофа ![]() получим выражение для изменения стандартной энергии Гиббса
получим выражение для изменения стандартной энергии Гиббса
![]() . (19.1)
. (19.1)
Тогда тепловой эффект реакции имеет вид
![]() . (19.2)
. (19.2)
Из уравнений (19.1) и (19.2) видно, что для определения ![]() и
и ![]() необходимо знать:
необходимо знать:
а) температурную зависимость ![]() , которая позволяет вычислять интеграл
, которая позволяет вычислять интеграл ![]() от 0 К до текущей температуры Т; б) тепловой эффект
от 0 К до текущей температуры Т; б) тепловой эффект 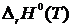 реакции при абсолютном нуле; в) при определении
реакции при абсолютном нуле; в) при определении ![]() необходимо знать постоянную интегрирования I. Величины
необходимо знать постоянную интегрирования I. Величины ![]() и
и ![]() относительно легко могут быть получены на основании ряда термических данных для реагирующих веществ.
относительно легко могут быть получены на основании ряда термических данных для реагирующих веществ.
Что же касается постоянной интегрирования I, то до открытия третьего закона термодинамики для её определения прибегали к экспериментальному определению стандартной константы равновесия исследуемой реакции хотя бы при одной температуре. Определив экспериментальным путем K0 при одной из температур, вычисляли ![]() , затем найденное значение
, затем найденное значение ![]() подставляли в уравнение (19.1) и тем самым получали возможность вычислять величину I.
подставляли в уравнение (19.1) и тем самым получали возможность вычислять величину I.
Если же величина 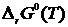 неизвестна ни при одной из температур, то в уравнении (19.1) I остаётся неопределённой константой. Проблема определения постоянной интегрирования I в уравнении (19.1), минуя экспериментальное исследование химического равновесия, привлекала вначале XX в. вВнимание многих физико-химиков. В частности, Ричардс при исследовании э. д.с. ряда гальванических элементов при различных температурах, вплоть до температуры жидкого воздуха, установил, что значения
неизвестна ни при одной из температур, то в уравнении (19.1) I остаётся неопределённой константой. Проблема определения постоянной интегрирования I в уравнении (19.1), минуя экспериментальное исследование химического равновесия, привлекала вначале XX в. вВнимание многих физико-химиков. В частности, Ричардс при исследовании э. д.с. ряда гальванических элементов при различных температурах, вплоть до температуры жидкого воздуха, установил, что значения ![]() и
и ![]() при низких температурах очень быстро сближаются друг с другом.
при низких температурах очень быстро сближаются друг с другом.
Нернст в 1906 г. в своей работе "О вычислении химического равновесия из термических данных" сформулировал постулат, согласно которому кривые в координатах 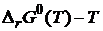 и
и 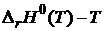 для любого химического процесса в конденсированных системах вблизи абсолютного нуля асимптотически приближаются друг к другу, т. е. имеют общую касательную при Т = 0 К. Причём для конденсированных систем индексы «P» и «0» можно не писать, так как для них
для любого химического процесса в конденсированных системах вблизи абсолютного нуля асимптотически приближаются друг к другу, т. е. имеют общую касательную при Т = 0 К. Причём для конденсированных систем индексы «P» и «0» можно не писать, так как для них 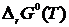 и практически не зависят от давления.
и практически не зависят от давления.
![]() (19.3)
(19.3)
Постулат Нернста и вытекающие из него следствия справедливы лишь для систем, состоящих из кристаллических веществ. Поэтому его часто называют теоремой Нернста (тепловой закон Нернста). Из данного постулата вытекает ряд важных следствий.
Следствие 1:
![]() (19.4)
(19.4)
Для доказательства (19.4) воспользуемся уравнением Гиббса — Гельмгольца, согласно которому
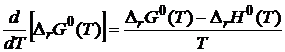 . (19.5)
. (19.5)
Так как при абсолютном нуле, как это следует из соотношений (19.1) и (19.2) 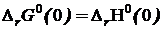 , то из (19.5) следует
, то из (19.5) следует
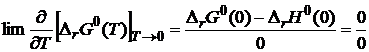
Для раскрытия неопределенности воспользуемся правилом Лопиталя:
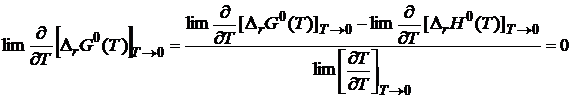
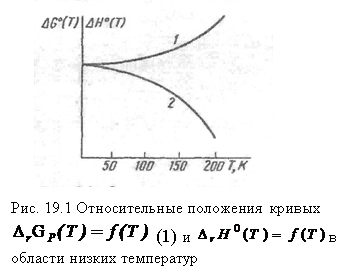 Таким образом, в соответствии с первым следствием касательная к кривым
Таким образом, в соответствии с первым следствием касательная к кривым 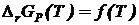 и
и 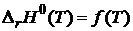 должна быть параллельной оси температур (см. рис. 19.1)
должна быть параллельной оси температур (см. рис. 19.1)
Следствие 2:
![]() . (19.6)
. (19.6)
Это следствие непосредственно вытекает из закона Кирхгофа ![]() и (19.4):
и (19.4):
![]() .
.
Из соотношения (19.6) следует, что процессы в конденсированных системах при температурах, близких к 0 К, протекают без изменения теплоёмкости. Отсюда следует, что теплоёмкости при T 0 К должны быть аддитивными величинами.
Как показывает опыт и квантовая теория, для кристаллов в отсутствие дефектов не только 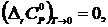 но и теплоёмкость каждого из компонентов системы при T→0 также стремится к нулю, т. е
но и теплоёмкость каждого из компонентов системы при T→0 также стремится к нулю, т. е
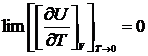 и
и 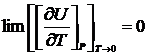 .
.
Следствие 3: для процессов с кристаллическими веществами
![]() , (19.7)
, (19.7)
где S0 — энтропия одного моля кристаллического вещества при 0K.
Из уравнения Гиббса-Гельмгольца при P=const и T=const имеем
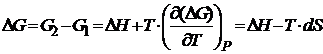 , из (19.4)
, из (19.4) 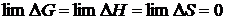 ,
,
тогда
![]() .
.
Следовательно, процессы в кристаллических веществах при температуре, близкой к 0 К, не сопровождаются изменением энтропии.
Поэтому энтропия, как и теплоёмкость при процессах между кристаллическими веществами при T→ 0 должна быть аддитивной величиной: только при этих условиях ![]() .
.
Следствие 4: для процессов с кристаллическими веществами постоянная интегрирования I в уравнении (19.1) равна 0.
Для доказательства запишем выражение для потенциала Гиббса
G=U-TS+PV=A+PV=H-TS
Комбинируя его с уравнениями
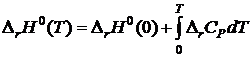
![]()
и 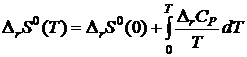 , получим
, получим
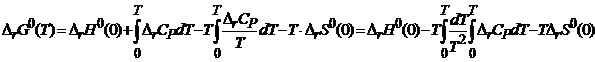 . (19.8)
. (19.8)
Из (19.1) и (19.8) следует, что ![]() , т. е.
, т. е.
![]() . (19.9)
. (19.9)
Изменение энтропии 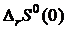 при процессах с кристаллическими веществами в соответствии с третьим следствием равно нулю. Постоянная I в (19.1) также должна быть равной нулю.
при процессах с кристаллическими веществами в соответствии с третьим следствием равно нулю. Постоянная I в (19.1) также должна быть равной нулю.
Итак, если все реагирующие вещества являются кристаллическими и бездефектными, то уравнение (19.1) переходит в
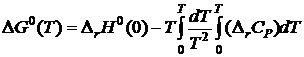 .
.
Следствие 5: для реакции ν1A1 + ν2A2 ↔ ν3A3 + ν4A4, протекающей в газовой фазе, постоянная интегрирования ![]() в уравнении равна алгебраической сумме истинных химических постоянных j:
в уравнении равна алгебраической сумме истинных химических постоянных j:
Значения истинных химических постоянных могут быть вычислены по экспериментальным данным зависимости давления насыщенною пара от температуры или методами статистической термодинамики. Поскольку для реакции в газовой среде ![]() , то
, то
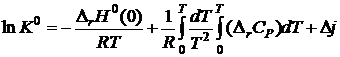 . (19.11)
. (19.11)
§2. Третий закон термодинамики
Таким образом, константу равновесия К0 в принципе можно вычислить используя только термические данные для реагирующих веществ.
Но в настоящее время уравнение (19.11) не используется при расчете константы равновесия, так как известны более простые и точные методы ;вычисления К0 при различных температурах. Большинство современных методов расчета химического равновесия базируется на третьем законе термодинамики, который можно сформулировать следующим образом
Энтропия любого индивидуального бездефектного кристаллического вещества при абсолютном нуле равна нулю
S(0) =
Этот закон в виде постулата сформулировал М. Планк в 1912 г.
Третий закон термодинамики может быть обоснован с помощью законов статистической термодинамики. Больцман доказал, что энтропия системы связана со статистическим весом соотношением
![]() . (19.13)
. (19.13)
Где k – постоянная Больцмана; Ω — статистический вес или термодинамическая вероятность данного макросостояния.
Под статистическим весом понимают число микросостояний, с помощью которых может быть реализовано данное макросостояние.
С точки зрения статистической теории энтропия идеального кристаллического вещества может быть равна нулю при 0 К только если Ω = 1, т. е. когда данное макросостояние может быть осуществлено только через одно микросостояние. Во всех остальных случаях энтропия кристаллического вещества при абсолютном нуле должна быть больше нуля,
Третий закон термодинамики позволяет вычислять так называемые абсолютные значения энтропии для любого вещества в любом агрегатном состоянии, если известны экспериментальные значения теплоёмкостей от 0 К до данной температуры, а также теплоты фазовых переходов.
Данным путем могут быть вычислены значения S0(298) для веществ при стандартных условиях. Другой путь определения стандартных энтропий основан на использовании спектроскопических данных о строении вещества. Значения S°(298) широко используются при вычислении изменения стандартной энергии Гиббса и стандартной константы химического равновесия.
Утверждение о том, что S(0)=0 нельзя распространять на твердые растворы. Для них при T = 0 появляется остаточная (нулевая) энтропия. В частности, для одного моля твердого раствора, если допустить, что он является бездефектным вплоть до абсолютного нуля, и если для каждого i-го компонента S(0)i =0, то при 0 K остаточная энтропия будет равна
S(0)=-RΣxi∙lnxi.
Например, для бинарного твердого раствора с х1 = x2 = 0,5 при абсолютном нуле S(0) = R∙1n2 = 5,76 Дж/(моль∙К).
Для твёрдого водорода остаточная энтропия при 0 К обусловливает существование двух его модификаций: пара - и орто-водорода. В связи с этим твёрдый водород также можно рассматривать как раствор (орто - и пара-водорода), энтропия которого не падает до нуля при 0 К. Наличие остаточной энтропии у СО (NO, N20) связано с различной ориентацией молекул СО в кристалле (ОС—СО и СО—СО). Так как атомы С и О близки по своим размерам, то эти два вида ориентации в кристалле должны обладать практически одинаковой энергией.
Отсюда статистический вес наинизшего энергетического уровня отдельной молекулы равен 2, а для моля кристалла — 2N. Поэтому остаточная энтропия СО должна быть величиной порядка R∙ln2 = 5,76 Дж/(моль∙К). Сравнение значений стандартной энтропии СО, вычисленных на основании калориметрических измерений [193,3 Дж/(моль∙К)] и спектроскопических данных [197,99 Дж/(моль∙К)], подтверждает этот вывод. Для твердых веществ, кристаллические решетки которых имеют какие-либо дефекты, S(0) ≠ 0. Значения остаточной энтропии отдельных веществ, как правило, небольшие величины по сравнению S0(298). Поэтому, если пренебречь остаточной энтропией (т. е. принять условно S(0) = 0), то это мало повлияет на точность термодинамических расчетов.
Кроме того, если учесть, что при термодинамических расчетах оперируют изменением энтропии при протекании процесса, то эти ошибки в значениях энтропии могут взаимно погашаться. Почти каждый химический элемент представляет собой смесь изотопов. Смешение изотопов, как и образование твердых растворов, ведет к появлению остаточной энтропии. Остаточная энтропия связана с ядерными спинами. Если учесть, что при протекании обычных химических реакций не изменяется изотопный состав системы, а также спины ядер, то остаточными составляющими энтропии при вычислении изменения энтропии ![]() можно пренебречь.
можно пренебречь.
Итак, третий закон термодинамики, согласно которому энтропия идеальных кристаллов при 0 К равна нулю, не является точным утверждением. Вычисленные на основания калориметрических данных Ср = f(Т) и теплот фазовых переходов так называемые абсолютные значения энтропии носят условный характер. Несмотря на отмеченные условности, третий закон термодинамики имеет большое практическое значение. На базе третьего закона термодинамики были разработаны современные методы вычисления изменения стандартной энергии Гиббса и константы равновесия при различных температурах.
§3. Расчет изменения стандартной энергии Гиббса и константы равновесия по методуТёмкина — Шварцмана
Для определения функциональной зависимости проинтегрируем уравнение Гиббса — Гельмгольца
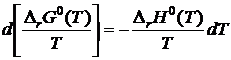
В пределах изменения температуры от 298,15 до T К:
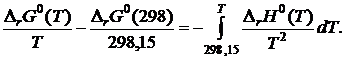 (19.14)
(19.14)
Согласно уравнению Кирхгофа
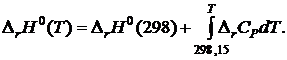 (19.15)
(19.15)
Подставляя ![]() из выражения (19.15) в уравнение (19.14), получим
из выражения (19.15) в уравнение (19.14), получим
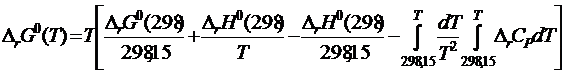 . (19.16)
. (19.16)
![]() . (19.17)
. (19.17)
После подстановки (19.17) в (19.16) будем иметь
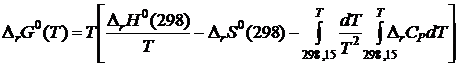 . (19.18)
. (19.18)
Если зависимость истинных молярных теплоемкостей от температуры для одной части компонентов реакции выражена степенными рядами вида
CP = a + bT + c’∙T-2,
а для другой части — рядами
CP = a + bT + c∙T2,
То изменение теплоемкости системы в результате реакции будет равно
ΔCP = Δa + Δb∙T + Δc∙T2 + Δ c’∙T-2.
Подставляя полученный степенной ряд для ΔСр в уравнение (19.18) и введя обозначение
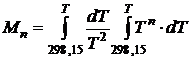 (19.19)
(19.19)
имеем
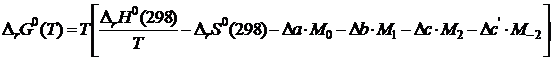 . (19.20)
. (19.20)
Подставляя выражение (19.20) в уравнение для ![]() , получим
, получим
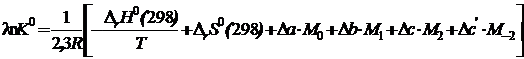 (19.21)
(19.21)
Для упрощения расчетов ΔG0(T) по уравнению (19.20) и ℓnK0 по уравнению (19.21) Тёмкиным и Шварцманом были вычислены интегралы Mn при различных температурах. Итак, при вычислении изменения энергии Гиббса по уравнению (19.20) и константы равновесия по уравнению(19.21) необходимо знать для каждого реагента: 1) температурную зависимость теплоёмкости Ср = f(T);
2) стандартную теплоту образования ![]() ; 3) стандартную энтропию S0(298).
; 3) стандартную энтропию S0(298).
§4. Расчёт изменения стандартной энергии Гиббса и константы равновесия с помощью функций приведённой энергии Гиббса
Под стандартным приведенным термодинамическим потенциалом (приведённой энергией Гиббса) понимают функцию
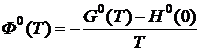 или
или 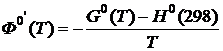 , (19.22)
, (19.22)
где 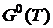 — стандартное значение энергии Гиббса при температуре T,
— стандартное значение энергии Гиббса при температуре T, 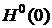 и
и
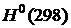 ; — стандартные значения энтальпии при 0 и 298 K соответственно.
; — стандартные значения энтальпии при 0 и 298 K соответственно.
Из соотношения 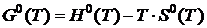 вытекает, что при Т = 0 и
вытекает, что при Т = 0 и ![]() . Числовые значения (G0(0) и H0(0) неизвестны. На практике обычно оперируют разностью G0(T) — G0(0) = G°(T) —H0(0), которая может быть определена экспериментальным путём.
. Числовые значения (G0(0) и H0(0) неизвестны. На практике обычно оперируют разностью G0(T) — G0(0) = G°(T) —H0(0), которая может быть определена экспериментальным путём.
В настоящее время функции Ф° и Ф0' вычислены при различных температурах с большой точностью для значительного числа газообразных веществ (состояние идеального газа) по молекулярным постоянным.
Для кристаллических веществ, подчиняющихся третьему закону термодинамики, расчёт Ф0 основан на использовании экспериментальных данных для теплоемкости ![]() от самой низкой температуры (≈ 4 K) до текущей температуры. Функция Ф0 находится из уравнения
от самой низкой температуры (≈ 4 K) до текущей температуры. Функция Ф0 находится из уравнения
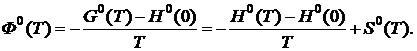
Так как 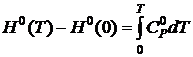 и
и 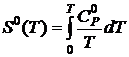 , то
, то
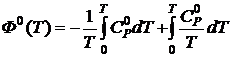 . (19.23)
. (19.23)
Вычисленные значения Ф°(Т) приведены в справочниках. Связь между приведенным термодинамическим потенциалом реагентов и изменением стандартной энергии Гиббса при реакции устанавливается следующим путем:
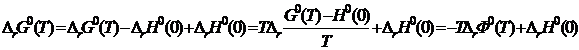 ,
,
где 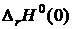 — стандартный тепловой эффект реакции при 0 К. Уравнение для расчета константы равновесия имеет вид
— стандартный тепловой эффект реакции при 0 К. Уравнение для расчета константы равновесия имеет вид
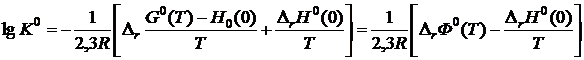 .(19.25)
.(19.25)
Для расчета ΔrG0(T) по уравнению (19.24) и lgK0 по уравнению (19.25) необходимо знать Ф° для каждого реагента при данной температуре и величину ΔrH°(0). Стандартный тепловой эффект при 0 К [ΔrH0(0)] может быть определен несколькими способами. В частности, ΔrH°(0) можно рассчитать по второму следствию из закона Гесса:
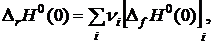 (19.26)
(19.26)
где [ΔfH0(0)]i — стандартная теплота образования при 0 К i-го реагента. Если стандартные теплоты образования при 0 К отсутствуют, то [ΔrH0(0)] можно вычислить на основании табличных данных для стандартных теплот образования [ΔrH0(298)] и высокотемпературных составляющих энтальпий [H0(T)-H0(298)] при 298 К для каждого реагента. Значение ΔrН°(0) рассчитывают по уравнению
![]() . (19.27)
. (19.27)
Если в качестве базисной температуры выбран не 0 К, а 298 К то уравнения (19.24) и (19.25) соответственно примут вид
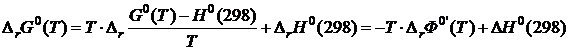 (19.28)
(19.28)
и 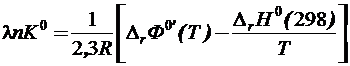 (19.29)
(19.29)
Значения высокотемпературных составляющих энтальпии [H0(T)-H0(298)] и функции 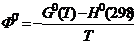 для большинства веществ определяются из справочников. Расчёт равновесия реакций с помощью приведённых термодинамических потенциалов Ф0 и Ф0’ широко используется на практике.
для большинства веществ определяются из справочников. Расчёт равновесия реакций с помощью приведённых термодинамических потенциалов Ф0 и Ф0’ широко используется на практике.

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 |
