

Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
1.3 Растворители
Многие процессы с межфазным переносом проводятся в отсутствии растворителя. Тем не менее, довольно обычным является проведение реакции, в присутствии органического растворителя или сорастворителя, особенно если субстрат твердый. Используемый в МФК растворитель не должен смешиваться с водой, так как в противном случае будут образовываться сильно гидратированные, «экранированные» ионные пары с низкой реакционной способностью. Чтобы избежать образования водородных связей с анионами ионных пар, растворитель, кроме того, должен быть апротонным. Чаще всего в качестве растворителя в процессах межфазного катализа в системах "жидкость-жидкость" и "жидкость-твердая фаза" используются бензол, гептан, толуол и другие углеводороды. Успешно применяются низкокипящие хлорированные углеводороды (хлороформ, дихлорметан), что объясняется их доступностью и легкостью удаления из реакционной смеси [2, 8]. Недостаток таких растворителей состоит в том, что они могут вызвать побочные реакции [12, 76, 77], однако это не очень опасно вследствие высокой скорости большинства реакций МФК [2, 8]. В системах "жидкость-твердая фаза" в качестве растворителя часто используют ацетонитрил, который нельзя применять в жидких двухфазных системах, поскольку он смешивается с водой [8].
До настоящего времени имеется только одно полное систематическое исследование влияния растворителей в МФК, проведенное французскими исследователями для системы «ж/ж». Они изучили 15 разнообразных растворителей, однако каких-либо общих выводов сделать не смогли [78].
Влияние растворителей на реакции нуклеофильного замещения в системе "жидкость-твердое тело" было изучено авторами работы [79]. В качестве объектов исследованиями было взято 17 растворителей, большинство из которых входит в список французских ученых. Никакой определенной связи между большинством физических параметров растворителей и скоростью реакции выявлено не было. Неожиданным фактом оказалось ускорение реакции в азотсодержащих высокоосновных растворителях без добавления катализатора. И чем меньше ускоряющее действие такого растворителя на скорость реакции, тем больше вклад катализатора. При прочих равных условиях реакция идет лучше при уменьшении диэлектрической проницаемости растворителя. Это соответствует предположению, что реакции МФК идут тем лучше, чем меньше степень диссоциации ионных пар. В то же время оказалось, что имеется хорошая корреляция между константой скорости реакции и таким чисто физическим параметром, как поляризуемость.
При таком анализе исследованные растворители распадаются на две группы, одна из которых включает все ароматические растворители и CCl4, а другая все остальные. Растворители первой группы проявляют в системе "жидкость-твердая фаза" так называемый "бензольный эффект". Он состоит в том, что бензол, несмотря на низкую диэлектрическую проницаемость, влияет на многие химические реакции так же сильно, как и полярные растворители [10]. Причиной "бензольного эффекта" считается высокая поляризуемость ароматического кольца [80, 81], а в случае CCl4 его объясняют возможным образованием комплексов с переносом заряда [82, 83].
Таким образом, в действии ароматических растворителей проявляется двойственность. С одной стороны, молекула растворителя воздействует как донорно-акцепторный агент, поляризуя химические связи в молекулах реагентов. Одновременно из-за наличия в молекулах растворителя ароматических систем возникает p-электронное взаимодействие с исходными и промежуточными частицами в ходе реакции, которое тем сильнее, чем больше "размыт" заряд в промежуточном комплексе.
Для ароматических растворителей характерна высокая поляризуемость и плохая сольватирующая способность. Азотсодержащие гетероциклы (типа пиридина) наряду с «ароматичностью» обладают и высокой сольватирующей способностью, поэтому являются «хорошими» растворителями. Очевидно, что при подборе растворителя необходимо учитывать как донорно-акцепторные характеристики, так и поляризуемость растворителя [10].
Таким образом, для того чтобы оценить возможность применения растворителя в каждом конкретном случае, необходимо иметь общее представление о его природе и типе проводимой реакции [8, 84].
1.4 Кинетика реакций в условиях МФК
Скорость химического превращения, протекающего в гетерофазной системе, должна зависеть от скорости перемешивания [13]. Перемешивание в таких системах изменяет величину поверхности раздела фаз и уменьшает диффузионное сопротивление со стороны сплошной фазы.
Если возможно неограниченное увеличение интенсивности перемешивания двухфазной системы, то поверхность раздела фаз при этом будет изменяться следующим образом. Вначале площадь поверхности S будет увеличиваться за счет уменьшения размера капель диспергированной фазы, так как отношение площади поверхности капли к ее объему обратно пропорционально радиусу (S/v » 1/r). Если мы будем продолжать дробить капли, то наступит момент, когда они станут соизмеримыми с размерами молекул или их ассоциатов и перестанут быть каплями [6].
Интенсивность перемешивания зависит от ряда факторов, таких как конструкция мешалки и геометрия реакционного сосуда [6, 13]. Исследования процессов перемешивания двух жидкостей привели к установлению эмпирического правила, связывающего величину поверхности раздела фаз с числом оборотов мешалки следующим образом:
∆S ~ wР, (1.9)
где р = 0,5-0,6 и зависит, главным образом, от конструкции мешалки [13].
Для проведения большинства лабораторных синтезов в условиях МФК можно использовать магнитную мешалку. Однако следует иметь в виду, что результаты не всегда являются хорошими, особенно в тех случаях, когда для реакции используют концентрированные растворы гидроксида натрия или калия, которые из-за их высокой вязкости перемешиваются недостаточно эффективно. Рекомендуются следующие скорости перемешивания: в нейтральных условиях в системе «вода-органическая фаза» — более 200 об/мин, для реакций в присутствии гидроксида натрия и в системах «твердая фаза-жидкость» — 750-800 об/мин. Для получения воспроизводимых кинетических данных необходимо проводить реакцию в условиях, когда скорость реакции не зависит от интенсивности перемешивания [36].
Для реакций с экстракционным механизмом МФК проявление плато на кривых зависимости скорости реакции от интенсивности перемешивания свидетельствует о том, что скорость диффузии переносимого реагента к ПРФ и вглубь диспергированной фазы заметно превосходит скорость химической реакции, что обеспечивает достижение экстракционного равновесия [13].
Появление плато на кривых зависимости скорости реакции от интенсивности перемешивания может быть объяснено двояким образом: либо достижением максимально возможной величины поверхности раздела фаз, либо тем, что скорость диффузии к поверхности раздела и вглубь диспергированной фазы обеспечивает достижение экстракционного равновесия [13]. При выполнении этого условия реакция практически полностью протекает в объеме фазы, и поэтому ее кинетика сходна с кинетикой обычных гомогенных реакций. В большинстве случаев стадия обмена анионами между органической и водной фазами характеризуется высокой скоростью (t1/2 ~ 1 с) и не проявляется в результатах кинетических измерений.
Для реакций с участием ПРФ, где одна из стадий реакции или сама реакция протекает на ПРФ, появление плато на кривых зависимости скорости реакции от интенсивности перемешивания обусловлено достижением максимально возможной величины ПРФ [36].
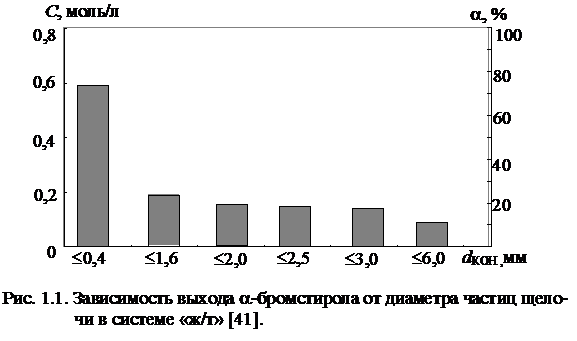
При изучении дегидробромирования 1,2-дибром-1-фенилэтана в двухфазных системах в присутствии бромида гексадецилтриметиламмония [41] установлено, что на активность системы «ж/т» существенное влияние оказывает увеличение степени дисперсности щелочи, что эквивалентно увеличению поверхности взаимодействия реагентов. Уменьшение размера частиц КОН в системе «ж/т» ведет к увеличению выхода основного продукта дегидробромирования 1,2-дибром-1-фенилэтана – a-бромстирола (рис. 1.1).
На рис. 1.2 представлены кривые зависимости выхода продукта дегидробромирования от интенсивности перемешивания в системах «ж/ж» и «ж/т».
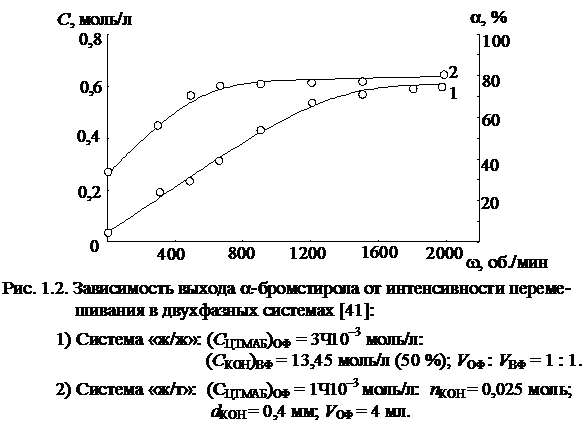
В системе «ж/ж» наблюдается выход на плато при 1200 об/мин, а в системе «ж/т» — при 500 об/мин В системе «ж/т» перемешивание предотвращает слипание (коагуляцию) частиц, а в системе «ж/ж» перемешивание обеспечивает процесс диспергирования с образованием эмульсии.
Выход на плато зависимости скорости реакции от интенсивности перемешивания связан с установлением максимальной поверхности раздела в результате равновесия между скоростью диспергирования и коалесценции.
1.5 Особенности межфазного катализа реакций, протекающих в присутствии оснований.
В присутствии сильных оснований проводят реакции различного типа, такие, как С-, О-, N- алкилирование, изомеризация, Н/D-обмен, присоединение, a-, b-элиминирование, гидролиз и многие другие. Важным преимуществом МФК в таких системах является возможность использования твердых гидроксидов щелочных металлов и их водных растворов, что исключает применение чувствительных к влаге и пожароопасных щелочных металлов, их алкоголятов, амидов и гидридов [2, 10, 50].
Механизм реакций с участием оснований. Херриотом и Пиккером [32] были опубликованы коэффициенты распределения аммониевых солей между раствором NaOH и бензолом, которые были получены ацидометрией аликвотных количеств фазы, содержащих Q+Х - в равновесии с NaOH. Эти данные поставили под сомнение возможность экстракции ионов ОН - в органическую фазу. Определение констант экстракции аммониевых гидроксидов (ЕQОН) довольно затруднительно вследствие наличия загрязнений в таких межфазных средах. Они могут быть внесены в процессе синтеза аммониевых солей, либо образованы в результате разложения катализатора под действием агрессивных сред. Так, для гидроксида тетрагептиламмония величина ЕQОН в бензоле равна 1,2. Гидроксид тетрабутиламмония, который часто используется в МФК/ОН--реакциях, вообще не способен экстрагироваться в бензол. Тем не менее, сотни МФК-реакций существенно ускоряются в присутствии такого гидрофильного катализатора как хлорид бензилтриэтиламмония, в то время как нуклеофильное замещение с этим катализатором по экстракционному механизму протекает медленно [11].
С тех пор как Макошей [12] была предложена гипотеза каталитического механизма для систем МФК/ОН-, а затем было показано, что константы экстракции для Q+ОН - в органических растворителях очень малы [2, 32], ывозможность реализации экстракционного механизма для обсуждаемых систем в научной литературе оценивается скептически. Но, несмотря на документированный факт плохой экстракции гидроксид-иона посредством Q+, все же появляются публикации, в которых успешность синтезов приписывается ионам ОН-, экстрагированным в органическую фазу [11].
Доказательством экстракционного механизма считается также независимость скорости реакции от скорости перемешивания при w > 300 об/мин. При незначительной скорости перемешивания (w < 300 об/мин) для данной реакции характерен период индукции, величина которого зависит от скорости диспергирования фаз и объясняется необходимостью установления стационарной концентрации Q+ОН-. Кроме того, скорость рассматриваемой реакции зависит от природы катализатора и противоиона [11].
Особенности, характерные для экстракционного механизма, отмечены в ряде работ, посвященных исследованию кинетики щелочного элиминирования, алкилирования и дейтерообмена. В частности, для реакций элиминирования механизм, включающий экстракцию Q+ОН - в органическую фазу приведен в работах [11, 85-87]. Доказательством механизма с участием ПРФ считаются:
– сложная кинетика (порядок реакции может быть дробным);
– существенное влияние интенсивности перемешивания фаз на скорость реакции [2, 11].
Несмотря на то, что модель межфазного механизма способна описать множество реакций, существует лишь небольшое количество кинетических работ в этой области, что подтверждает сложность этого феномена [11].
Физико-химические особенности МФК/ОН–-реакций. Самое существенное химическое свойство концентрированных водных растворов щелочей связано с высоким сродством свободных гидроксид-ионов к протонам. Это позволяет проводить депротонирование различных органических соединений в системах «ж/ж» и получать карбанионы — высокореакционные промежуточные соединения, вступающие в дальнейшие превращения. Гидроксид-ион является одним из самых сильных оснований: среди одновалентных ионов он уступает только амид-иону. Вода обладает несравненно более слабыми основными свойствами [6].
С повышением концентрации щелочи, степень ее диссоциации уменьшается незначительно. Так, даже в 48 %-ном водном растворе диссоциировано не менее 71 % всех молекул. Поэтому можно считать, что активной частицей в концентрированном растворе щелочи является OH-. Возрастание коэффициента активности гидроксд-иона (gOH) и снижение активности воды (aW) [88] объясняется образованием сильной водородной связи между ионом ОН- и тремя молекулами воды [6].
Следует отметить, что в таких системах также проявляется различие калиевой и натриевой щелочи: хотя характер кривых зависимости gOH от концентрации для обеих щелочей подобен, в близкой к насыщению области они заметно различаются. Возможно, это обстоятельство связано с уменьшением количества «свободной воды».
В любом случае чрезвычайно низкая активность воды в 50 %-ных растворах щелочей вполне удовлетворительно объясняет, почему такие растворы ведут себя аналогично твердой щелочи, и их можно использовать для проведения реакций, требующих безводных условий (например, получение енолятов и пр.), или реакций, в которых участвуют крайне чувствительные к влаге реагенты (такие, как неустойчивые илиды фосфора или серы) [9].
Растворимость органических веществ в концентрированных растворах щелочей весьма мала. В ряде случаев растворение органического вещества включает также химическое взаимодействие со щелочью, проявляющееся, например, в образовании енолятов. Так, с водой ацетон смешивается неограниченно, но добавление щелочи приводит к его «высаливанию» из водной фазы; уже в 28 %-ном растворе NaOH растворимость ацетона не превышает 1,1 % [6].
Низкая растворимость органических веществ в концентрированных растворах щелочей отличает эти растворы от других водных фаз, используемых в двухфазных каталитических системах. Растворимость щелочей в органической фазе также незначительна. Даже в присутствии межфазного переносчика экстракция Q+ОН– в органическую фазу очень невелика [2, 11, 32].
Существует еще одна проблема, связанная с применением концентрированных растворов щелочей. В двухфазных системах плотности органической и водной фаз сильно отличаются друг от друга. Поэтому необходимым условием при изучении механизма реакций в двухфазных системах является обеспечение интенсивного перемешивания водной и органической фаз. Перемешивание в этих системах может представлять проблему, так как растворы становятся вязкими и склонны давать эмульсии. Выходы и скорости в случае, если не удается добиться идентичных условий перемешивания, обычно не воспроизводимы [2].
Наиболее простым методом получения негидратированных ("голых") анионов ОН– является их перевод из твердой фазы в несольватирующий растворитель. В этом случае гидратной воды просто нет, и желаемый результат достигается самым прямым способом. Однако в системах «ж/т», содержащих твердую щелочь, образование воды на стадии депротонирования приводит к гидратации гидроксид-иона. Система «ж/т» с твердой щелочью менее изучена, чем система с концентрированным водным раствором щелочи, однако в ряде случаев активность систем «ж/т» существенно выше [41, 39].
2. Реакции элиминирования галогеноводорода в условиях межфазного катализа.
Применение МФК для реакций элиминирования позволяет решить ряд проблем и увеличить выход целевого продукта даже мягких условиях проведения реакции. МФК открывает наиболее реальную возможность усовершенствования технологии дегидрогалогенирования и в целом ряде случаев может сделать его вполне конкурентоспособным и более привлекательным, чем соответствующие высокотемпературные процессы.
По своему значению в органическом синтезе дегидрогалогенирование представляет собой наиболее важную группу реакций b-элиминирования. Однако работ, посвященных исследованию количественных закономерностей таких реакций, крайне мало [87].
Как указано в [6], соотношение между экспериментальными и теоретическими работами по МФК составляет приблизительно 1 : 7. Та же тенденция, пожалуй, лишь еще более ярко выраженная, верна и для реакций дегидрогалогенирования, проводимых в условиях МФК [87].
2.1 Механизм элиминирования
В настоящее время нет единого мнения о механизме МФК в присутствии щелочей и, в частности, о механизме реакций элиминирования. На механизм реакций оказывают влияние, нередко противоречивое, много факторов, что не позволяет включить их в какую-либо стройную теорию [40].
По мнению авторов работы [89], главное препятствие в успешном применении МФК для реакций b-элиминирования галогенводородов заключается в самой природе каталитических процессов, а именно в ионообменном равновесии. Высокая эффективность генерирования карбанионов в присутствии водного раствора гидроксида натрия и межфазного катализатора (обычно используются тетраалкиламмониевые соли Q+X-) зависит от механизма катализа [89].
В том случае, если реализуется механизм МФК с участием ПРФ, основная стадия в этих каталитических процессах — отщепление протона — происходит на границе раздела органической фазы и раствора щелочи. Полученные карбанионы, локализованные на границе раздела фаз, вступают в ионообменные процессы с катализатором с образованием липофильных ионных пар, которые далее мигрируют в органическую фазу, где и происходит основная реакция [11, 12, 89]. Тогда образование  и их переход в органическую фазу в соответствии с уравнением:
и их переход в органическую фазу в соответствии с уравнением:
Q+X–о. ф. + NaOHв. ф. ↔ Q+OH–о. ф. + NaX в. ф. (1.10)
не влияет на стадию каталитического генерирования карбанионов и поэтому этот процесс не должен зависеть от ионообменного равновесия.
Однако с другой стороны, реакцию межфазно-каталитического элиминирования, можно представить следующей схемой:

H2Oо. ф. ↔ H2Oв. ф. , (1.12)
согласно которой анионы OH - должны экстрагироваться в органическую фазу в виде ионных пар Q+OH-. В этом случае процесс, представленный уравнением (1.10) сильно зависит от ионообменного равновесия, которое будет определять концентрацию ионов OH - в органической фазе [11, 85, 86].
По мере протекания реакции элиминирования, концентрация ![]() увеличивается в соответствии с ур. (1.11), а концентрация
увеличивается в соответствии с ур. (1.11), а концентрация ![]() в органической фазе становится незначительной, и как следствие, элиминирование прекращается. Авторы [1, 89] считают, что b-элиминирование может быть успешно реализовано в условиях ионно-парного экстрагирования, т. к. в этом случае обеспечиваются эквимолярные количества Q+OH-, получаемые ионным обменом между раствором щелочи и гидросульфатом тетрабутиламмония.
в органической фазе становится незначительной, и как следствие, элиминирование прекращается. Авторы [1, 89] считают, что b-элиминирование может быть успешно реализовано в условиях ионно-парного экстрагирования, т. к. в этом случае обеспечиваются эквимолярные количества Q+OH-, получаемые ионным обменом между раствором щелочи и гидросульфатом тетрабутиламмония.
Однако преимущества МФК-процессов могут быть утеряны, если дорогостоящие ониевые соли использовать в стехиометрических количествах [89, 90]. Тем не менее, известны многочисленные примеры реакций элиминирования с использованием каталитических количеств ЧАС [12, 8, 11, 85-87], причем некоторыми авторами [85, 86] предложен механизм экстракции для реакций дегидробромирования. Не отвергается такая возможность и в ряде других публикаций [11, 87]. В [41] при изучении кинетики и механизма реакции дегидробромирования 1,2-дибром-1-фенилэтана получены экспериментальные доказательства протекания реакции на ПРФ как в системах «ж/ж», так и в системах «ж/т».
2.2 Влияние добавок органических кислот, органических оснований и воды.
В работе [2] отмечается, что дегидрогалогенирование в присутствии гидроксидов щелочных металлов проходит только в тех случаях, когда в соединении имеется протон, активированный электроноакцепторным заместителем или находящийся в бензильном или винилогичном к бензильному положении. Круг субстратов, способных дегидрогалогенироваться в условиях МФК, можно расширить за счет использования четвертичных аммониевых солей и кислотно-основных добавок [50].
Кислотно-основные добавки. В ряде работ для реакций элиминрования в условиях МФК рекомендуется использовать добавки органических соединений кислотно-основной природы — спиртов [89, 91-93], диолов [890], фенолов, карбоновых кислот [66], аминов [67], которые сами по себе, как правило, не являются переносчиками ионов из одной фазы в другую.
По мнению ряда исследователей [89, 91] возможность использования кислотно-основных добавок в реакциях элиминирования является одним из возможных путей решения проблемы плохой экстракции Q+OH - в органическую фазу. Органическая кислота Y-H в двухфазной системе образует липофильный анион Y![]() , который в свою очередь является основанием [89].
, который в свою очередь является основанием [89].
Анион Y![]() в составе ионной пары
в составе ионной пары ![]() идет в органическую фазу, где он направляет элиминирование на образование алкена, Y-H и Q+OH-. Последующее депротонирование Y-H на границе раздела фаз приводит к регенерированию аниона
идет в органическую фазу, где он направляет элиминирование на образование алкена, Y-H и Q+OH-. Последующее депротонирование Y-H на границе раздела фаз приводит к регенерированию аниона![]() и цикл, представленный уравнением 1.15 замыкается.
и цикл, представленный уравнением 1.15 замыкается.
Y–Hо. ф. + NaOHв. ф. ↔ Y–Na+ПРФ + H2Oв. ф. (1.13)
Y–Na+ПРФ + Q+X–о. ф. ↔ Q+Y–ПРФ + Na+X–в. ф. (1.14)
![]()
Для реакций элиминирования, проводимых в условиях сокатализа, предложена общая схема МФК-цикла [91] (схема 1.5):
Схема 1.5

Аналогичного мнения придерживаются Демлов и Тизер [90], исследовавшие влияние диолов в синтезе ацетиленовых соединений.
Влияние добавок спиртов в реакции дегидрохлорирования дихлорэтана в присутствии 50 %-ного раствора NaOH и тетраалкиламмониевых солей изучалось в [92], однако авторы этой работы рассматривают спирт как промотирующую добавку.
Предложенный механизм промотирующего действия спиртов для межфазных реакций под действием оснований не предусматривает фазового переноса ни ионных пар, ни ионов, что принципиально отличает его от каталитического цикла Макоши (схема 1.6) [92]:
Схема 1.6
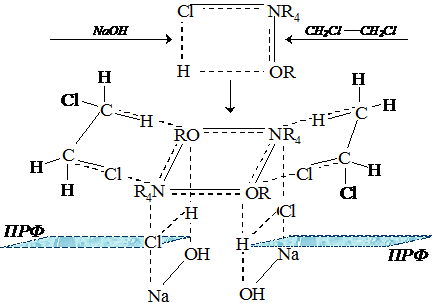
Предполагается, что катализ включает стадию образования ассоциата, его сольватацию с хлорорганическим субстратом и координацию образующегося агрегата с NаOH. Реакция идет в органической фазе и на поверхности раздела фаз [92].
Высказано предположение [41], что активирующее действие добавок липофильных спиртов в реакции дегидробромирования 1,2–дибром-1-фенилэтана в условиях МФК может быть обусловлено целым рядом причин, для подтверждения или исключения которых необходимы дальнейшие экспериментальные исследования:
1. Липофильные спирты депротонируются на ПРФ, а образующиеся алкоголят-ионы в качестве основания переносятся липофильным катионом в объем органической фазы, где происходит их взаимодействие с субстратом (гипотеза Макоши)
2. В условиях МФК небольшие количества спирта способствуют стабилизации Е1сВ-подобного переходного состояния механизма Е2. В случае 1,2-дибром-1-фенилэтана молекулы спирта специфически сольватируют атомы брома, усиливая их акцепторные свойства и увеличивая-I-эффект (без существенного гетеролиза связи С─Br в неполярном растворителе —толуоле). Это может приводить к увеличению подвижности a-водородного атома и способствовать образованию Е1сВ-подобного переходного состояния с карбанионным характером. Кроме того, активация молекул a-бромстирола добавками спирта способствует его дегидробромированию с образованием фенилацетилена.
3. Липофильные спирты могут влиять на эффективность межфазного катализа, так как обладают свойствами со-ПАВ по отношению к классическим ПАВ. Для смеси ПАВ и со-ПАВ гидрофильно-липофильный баланс (ГЛБ) является аддитивной величиной, и роль липофильного спирта в условиях МФК может быть обусловлена смещением ГЛБ в сторону липофильности. При этом в системах «ж/ж» облегчается диспергирование щелочной водной фазы с образованием обратных эмульсий «в/м» с более высокоразвитой поверхностью, а в системах «ж/т» на ПРФ образуется более липофильная «омега»-фаза с повышенной способностью к солюбилизации органического субстрата.
Влияние добавок воды. Имеющиеся в литературе данные по влиянию воды на скорость и механизм межфазных реакций в системах «ж/т» неоднозначны и противоречивы. В работе [94] показано, что в одном случае добавки воды приводят к уменьшению скорости процесса, в другом — к более чем двукратному ее увеличению, в третьем случе зависимость скорости процесса от количества воды имела экстремальный характер. Так, малые добавки воды резко увеличивают скорость реакции додецилхлорида с твердыми солями муравьиной кислоты при катализе метилтриоктиламмоний хлоридом в системе "о-дихлорбензол - твердая соль" [95]. Напротив, реакции алкилбромидов с КCl, проведенные в аналогичных условиях, нечувствительны к добавкам воды. Эти различия Юфит [10] связывает с прочностью кристаллической решетки (ПРК) реагента. В случае плоских ионов типа формиата с малой ПКР выявлено резкое увеличение скорости (и выхода) реакции при добавлении малых количеств воды, а для сферических ионов галогенидов с высокой ПКР малые количества добавленной воды никак не влияют на скорость реакции. Сферическая симметрия иона С1- позволяет создать крайне выгодный циклический комплекс, в котором катализатор не только координирует реагирующие молекулы, но и сольватирует уходящую группу. Необходимости гидратировать уходящую группу в этом случае нет, что проявляется в малой чувствительности реакции к добавлению воды [10].
Другой важной причиной разнонаправленного действия воды на скорость МФК-реакций является различие в механизмах межфазного взаимодействия.
В случае экстракционного механизма с быстро устанавливающимся равновесием распределения ионного реагента и катализатора определяющую роль играет степень гидратации аниона Y-, перенесенного в органическую фазу в виде ионной пары с катализатором Q+Y - [7]. Влияние гидратации на реакционную способность особенно значительно проявляется для высоко гидрофильных анионов ОН - [96-98] и F - [99, 100]. Установлено [101, 102], что увеличение числа гидратации фторида тетрагексиламмония от 0 до 3 заметно снижает скорость реакции замещения в октилметансульфонате, которая стремится к постоянной величине при дальнейшем увеличении степени гидратации. Этот факт позволяет предположить, что в случае лимитирующей скорость химической реакции в органической фазе наступает такая ситуация, когда добавки воды не влияют на скорость процесса. С одной стороны, это может происходить вследствие насыщения органической фазы водой, а с другой, по причине достижения предельной степени гидратации аниона [101].
Иная картина наблюдается в случае межфазных реакций в системах «ж/т», в которых процессы переноса через ПРФ или реакции на ПРФ вносят основной вклад в общую скорость расходования субстрата. При этом роль воды состоит в уменьшении прочности кристаллической решетки твердой фазы за счет гидратации ионных частиц [99, 103]. По мере увеличения содержания воды возможна смена скорость лимитирующей стадии до начала процесса химического взаимодействия, как это наблюдалось в работе [7] при изучении влияния добавок воды на скорость реакции бензилгалогенидов с твердым цианидом калия.
Для большинства реакций, протекающих в условиях МФК в системе «ж/т», влияние добавок воды на скорость реакции все же существенно и носит экстремальный характер [2, 7, 8, 94, 99-105], причем действие воды наиболее эффективно в области низких концентраций. Это явление авторы [7, 69, 101] связывают с образованием на поверхности твердой фазы своеобразной водной пленки «омега»-фазы, в которой концентрируется межфазный катализатор и реализуются благоприятные условия для образования реакционного комплекса с участием катализатора, аниона и субстрата.
В работе [41] показано, что при дегидробромировании 1,2-дибром-1-фенилэтана в условиях МФК в системе «ж/т» оптимальное содержание воды составляет 1,6 мол. % от количества щелочи (рис 1.3).

2.3 Субстраты для реакций элиминирования
Природа отщепляющегося вещества в реакциях элиминирования может быть разнообразной: вода, аммиак, амины, галогены и т. д. Сегодня метод МФК применяется для реакций a-, b-, g-элиминирования, многие из которых имеют препаративное и промышленное значение.
Одной из самых широко распространенных межфазно-каталитических реакций a-элиминирования является реакция образования дигалогенкарбенов [2, 6]. Общепринятым методом получения дигалогенкарбена является его образование из хлороформа [2, 12].
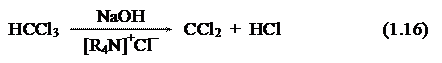
К реакциям g-элиминирования в условиях МФК относятся реакции получения ангидридов и фторангидридов кислот. В работе [2] описан метод получения алифатических и ароматических фторангидридов в условиях МФК из соответствующих кислот, карбоната калия и димера гексафторпропена, перфтор-2-метил-пентена-2 или его тримеров в дихлорметане.
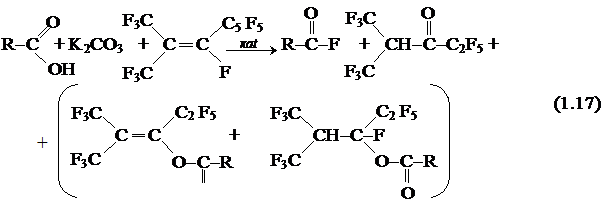
В практическом отношении наиболее важное значение имеют реакции b-элиминирования. Как и в гомогенных средах, в условиях МФК элиминирование конкурирует с реакциями нуклеофильного замещения. Отщепление становится преобладающим при использовании вторичных субстратов (особенно циклогексильные производные). Третичные соединения дают олефины в качестве единственного продукта [2].
МФК был успешно применен в синтезе алкенов из полигалогеналканов элиминированием галогенов под действием цинка в водной среде [39]:
![]() (1.18)
(1.18)
Довольно большую и, пожалуй, самую важную группу составляют реакции дегидрогалогенирования. Не случайно, что первые работы в этой области связаны именно с использованием МФК в ряде промышленно важных процессов. Большой интерес представляет синтез 2,3-дихлорбутадиена-1,3 — ценного мономера для получения сополимеров с хлоропреном и другими мономерами, обладающих комплексом ценных свойств. Получение 2,3-дихлорбутадиена-1,3 в условиях МФК выгодно отличается от других известных способов простотой, доступностью исходного сырья и технологичностью [50, 87].
Одним из важных достижений МФК является возможность синтеза ацетиленовых соединений. Сначала синтез ацетиленовых производных проводили в присутствии стехиометрических количеств тритона В (гидроксида бензилтриметиламмония). Доступный продажный продукт —раствор этой щелочи в метаноле —был превращен в бензольные, толуольные или пиридиновые растворы путем разбавления метанольного раствора избытком нового растворителя и отгонки большей части метанола. Тритон В не всегда полностью растворяется в бензоле или толуоле, однако, в большинстве случаев это не имеет значения. Типичный пример такой реакции приведен ниже [2].
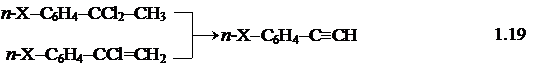 В настоящее время ацетиленовые производные получают в истинных условиях МФК, т. е. с использованием не стехиометрических, а каталитических количеств солей тетраалкиламмония и других межфазных переносчиков. В табл. 1.2 представлены некоторые примеры межфазных систем, в которых наблюдалось элиминирование до алкинов [8].
В настоящее время ацетиленовые производные получают в истинных условиях МФК, т. е. с использованием не стехиометрических, а каталитических количеств солей тетраалкиламмония и других межфазных переносчиков. В табл. 1.2 представлены некоторые примеры межфазных систем, в которых наблюдалось элиминирование до алкинов [8].
Таблица 1.2
Элиминирование под действием нуклеофилов: синтез алкинов [8].
Реакция | Реагент/катализатор | Выход, % |
Cl2C=CHCl ® СlCºССl | Q+Cl-/эпихлоргидрин | 75 |
CH2BrCHBrCH(OC2H5)2 ® ® HCºCCH(OC2H5)2 | NaOH/BuN+Br- | 80 |
NCCCl=C(CO2CH3)2 ® ® NCCºCCOOCH3 | KCl/дицикло-18-краун-6 | 16 |
C6H5CHBrCH2Br ® ® C6H5CºCH | NaOH/BuN+Br | 87 |
CH3OCOCCl=C(CO2CH3) ® ® CH3OCOCºCCO2CH3 | KCl/ дицикло-18-краун-6 | 7 |
C6H5CHBrCHBrC6H5 ® ® С6Н5СºСС6Н5 | NaOH/BuN+Br | 75 |
Необходимо упомянуть еще об одном достоинстве МФК — возможности проведения элиминирования совместно с другими реакциями. Так, разработан способ получения дипропаргилового эфира из трибромпропана. Этот процесс включает четыре стадии, которые благодаря применению МФК осуществляются в одном реакторе без выделения и очистки промежуточных соединений [40].

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 10 11 |
