

Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
ГОСУДАРСТВЕННОЕ ОБРАЗОВАТЕЛЬНОЕ УЧРЕЖДЕНИЕ
ВЫСШЕГО ПРОФЕССИОНАЛЬНОГО ОБРАЗОВАНИЯ
«ТЮМЕНСКИЙ ГОСУДАРСТВЕННЫЙ УНИВЕРСИТЕТ»
На правах рукописи
МАМОНТОВА Юлия Владимировна
СИНЕРГИЗМ ДЕЙСТВИЯ КАТАЛИЗАТОРОВ МЕЖФАЗНОГО ПЕРЕНОСА И СОЕДИНЕНИЙ КИСЛОТНО-ОСНОВНОЙ ПРИРОДЫ В РЕАКЦИИ ДЕГИДРОБРОМИРОВАНИЯ 1,2-ДИБРОМ-1-ФЕНИЛЭТАНА В ДВУХФАЗНЫХ СИСТЕМАХ
Специальность 02.00.03 – Органическая химия
ДИССЕРТАЦИЯ
на соискание ученой степени кандидата
химических наук
Научный руководитель: доктор педагогических наук,
кандидат химических наук, доцент
Тюмень - 2006
ВВЕДЕНИЕ. 6
I. ЛИТЕРАТУРНЫЙ ОБЗОР. 10
1. Современные представления о методе межфазного катализа. 10
1.1 Типы гетерофазных систем. 11
1.2 Межфазные катализаторы и механизм их действия. 13
1.3 Растворители. 22
1.4 Кинетика реакций в условиях МФК. 25
1.5 Особенности межфазного катализа реакций, протекающих в присутствии оснований. 28
2. Реакции элиминирования галогеноводорода в условиях межфазного катализа. 32
2.1 Механизм элиминирования. 32
2.2 Влияние добавок органических кислот, органических осно-ваний и воды. 34
2.3 Субстраты для реакций элиминирования. 40
II. ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ. 43
II.1. Реагенты и подготовка их к синтезу. 43
2.1. Синтез 1,2-дибром-1-фенилэтана. 44
2.2. Синтез a-бромстирола. 45
2.3 Синтез фенилацетилена. 46
2.4 Синтез b-бромстирола. 46
II.3. Методика каталитического дегидробромирования 1,2-дибром-1-фенилэтана. 47
3.1 Исследование влияния природы межфазных катализаторов на эффективность реакции дегидробромирования 1,2‑дибром-1-фенилэтана. 48
3.2 Исследование влияния концентрации межфазного катали-затора на эффективность реакции дегидробромирования 1,2‑ди-бром-1-фенилэтана. 48
3.3 Исследование влияния синергизма действия различных по природе межфазных катализаторов на реакцию дегидробромиро-вания 1,2-дибром-1-фенилэтана. 49
3.4 Исследование влияния соотношения двух межфазных ката-лизаторов в смеси на эффективность дегидробромирования 1,2-дибром-1-фенилэтана. 50
3.5 Исследование влияния карбоновых кислот на эффективность реакции дегидробромирования 1,2-дибром-1-фенилэтана в отсут-ствии межфазного катализатора. 50
3.6 Исследование влияния фенолов на эффективность реакции дегидробромирования 1,2-дибром-1-фенилэтана в отсутствии межфазного катализатора. 51
3.7 Исследованин влияния спиртов на эффективность реакции дегидробромирования 1,2-дибром-1-фенилэтана в отсутствии межфазного катализатора. 52
3.8 Исследование влияния аминов на эффективность реакции дегидробромирования 1,2-дибром-1-фенилэтана в отсутствии межфазного катализатора. 53
3.9 Определение оптимального соотношения межфазного катализатора и спирта для получения композиций, проявляющих синергетический эффект. 53
3.10 Исследование влияния совместного действия солей тетра-алкиламмония и карбоновых кислот на эффективность реакции дегидробромирования 1,2-дибром-1-фенилэтана. 54
3.11 Исследование влияния совместного действия солей тетраалкиламмония и фенолов на эффективность реакции дегидробромирования 1,2-дибром-1-фенилэтана. 55
3.12 Исследование влияния совместного действия солей тетра-алкиламмония и спиртов на эффективность реакции дегидро-бромирования 1,2-дибром-1-фенилэтана. 56
3.13 Исследование влияния совместного действия солей тетра-алкиламмония и аминов на эффективность реакции дегидро-бромирования 1,2-дибром-1-фенилэтана. 56
3.14 Исследование влияния концентрации субстрата на эффек-тивность реакции дегидробромирования 1,2-дибром-1-фенил-этана. 57
3.15 Исследование влияния температуры на эффективность реакции дегидробромирования 1,2-дибром-1-фенилэтана. 58
3.16 Получение кинетических кривых образования a‑бром-стирола и фенилацетилена в присутствии каталитических доба-вок 2-метилбутанола-2. 59
II.4. Методы анализа. 59
III. эксПЕРИМЕНТАЛЬНЫЕ РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ. 61
III.1 Эффективность межфазных катализаторов различной при-роды и синергизм их действия в реакции дегидробромирования 1,2-дибром-1-фенилэтана в системах «ж/ж» и «ж/т». 61
1.1 Влияние природы межфазных катализаторов и их липофиль-ности на эффективность реакции дегидробромирования. 63
1.2 Синергизм действия смесей различных по природе межфаз-ных катализаторов. 72
III.2 Каталитическая активность соединений кислотно-основной природы и синергизм действия их смесей с катализаторами межфазного переноса в реакции дегидробромирования 1,2-дибром-1-фенилэтана в системах «ж/ж» и «ж/т». 80
2.1 Влияние карбоновых кислот, фенолов и спиртов на эффек-тивность дегидробромирования в системах «ж/ж» и «ж/т» в отсутствие межфазного катализатора. 81
2.2 Влияние аминов на эффективность дегидробромирования в системах «ж/ж» и «ж/т» в отсутствие межфазного катализатора. .. 88
2.3 Определение оптимального соотношения межфазного ката-лизатора и спирта для получения синергетических композиций. .. 90
2.4 Синергизм действия солей тетраалкиламмония и соединений кислотно-основной природы. 92
2.5 Дегидробромирование 1,2-дибром-1-фенилэтана по второй стадии с образованием фенилацетилена. 110
IV. Выводы.. 114
Литература.. 116
ВВЕДЕНИЕ
Метод межфазного катализа (МФК) широко используется для проведения и регулирования реакций органического и нефтехимического
синтеза.
В последнее время значительно возрос интерес к двухфазным системам, содержащим щелочную фазу (водную или твердую), для проведения органических реакций элиминирования, а также других реакций, включающих стадию депротонирования субстрата. Особую привлекательность применение метода МФК (МФК/ОН--систем) имеет для повышения эффективности и селективности реакций дегидрогалогенирования галогенпроизводных углеводородов — основного способа получения алкенов и алкинов, являющихся ценным сырьем для органического синтеза разнообразных органических соединений. Важным преимуществом метода МФК в таких системах является также использование твердых гидроксидов щелочных металлов и их водных растворов, что позволяет исключить применение высокочувствительных к влаге и пожароопасных щелочных металлов для получения их алкоголятов, амидов и гидридов, позволяет избежать тщательной осушки реагентов и использования дорогостоящих апротонных растворителей.
В настоящее время установлено, что в МФК/ОН--системах дегидрогалогенирование субстратов происходит по механизму МФК с участием поверхности раздела фаз (ПРФ), что обусловлено неспособностью большинства известных катализаторов межфазного переноса эффективно экстрагировать высоко гидрофильный гидроксид-анион в объем органической фазы. Специфическое влияние ПРФ на механизм реакции элиминирования ограничивает возможность применения этого метода для субстратов, не имеющих активированного протона.
Для решения проблемы плохой экстракции гидроксид-ионов и обеспечения возможности протекания химической стадии процесса дегидрогалогенирования субстратов в объеме органической фазы представляет интерес использование смесей межфазных катализаторов или добавок сокатализаторов кислотно-основной природы (карбоновых кислот, фенолов, спиртов, аминов), однако, представления о механизме их синергетического действия и способах разработки наиболее оптимальных композиций до настоящего времени не выходят за рамки предположений и гипотез.
Сравнительное исследование активности катализаторов межфазного переноса различной природы, влияния кислотно-основных добавок на эффективность реакций элиминирования в системах "жидкость/жидкость" и "жидкость/твердая фаза" представляет значительный теоретический интерес для развития модельных представлений о механизме реакций в МФК/ОН--системах и о механизме синергетического действия различных катализаторов. Такие исследования также представляют большой практический интерес, связанный с необходимостью расширения круга субстратов и повышения эффективности и селективности процессов синтеза непредельных соединений.
Целью работы является исследование эффективности катализаторов межфазного переноса и их смесей с соединениями кислотно-основной природы в реакции дегидробромирования 1,2-дибром-1-фенилэтана для двух типов двухфазных систем:
а) "ж/ж" (органическая фаза + водный раствор щелочи),
б) "ж/т" (органическая фаза + твердый гидроксид щелочного металла).
Научная новизна. В работе впервые для реакций элиминирования, протекающих по механизму МФК с участием ПРФ, предложен принцип подбора наиболее эффективных катализаторов межфазного переноса ОН--ионов — солей тетраалкиламмония, — основанный на увеличении их каталитической активности с повышением липофильности в системах «ж/т» и увеличением поверхностной активности на границе раздела фаз в системах «ж/ж». Показано, что в системах «ж/т» эфиры бензо-15-краун-5 и ПЭГ могут проявлять свойства оснований в объеме органической фазы.
Впервые установлено наличие синергизма действия для двухкомпонентных смесей: "липофильная соль тетраалкиламмония (ТОАБ, ДДТМАБ и ГДТМАБ) – эфир (бензо-15-краун-5, ПЭГ-400)" и "липофильная соль тетраалкиламмония (ТОАБ, ДДТМАБ и ГДТМАБ) – соединение кислотно-основной природы (карбоновые кислоты, фенолы, спирты, амины), растворимое в органической фазе" для реакции дегидробромирования 1,2-дибром-1-фенилэтана в системах «ж/ж» и «ж/т» и предложено обоснование синергизма действия катализатора межфазного переноса и кислотно-основного сокатализатора, включающее возможность протекания реакции дегидробромирования субстрата при взаимодействии с основной формой сокатализатора в объеме органической фазы, а не на ПРФ, а также эффективность нейтрализации кислотной формы сокатализатора щелочью за счет межфазного переноса ионов ОН− катионами Q+ из щелочной водной или твердой фазы на ПРФ. Впервые установлено, что локализация химической стадии процесса в объеме органической фазы, а не на ПРФ, обеспечивает возможность дегидрогалогенирования субстратов, не имеющих активированного протона, за счет благоприятных условий для реализации переходного состояния механизма Е2 для реакций элиминирования.
Показано, что наиболее активные смеси, содержащие липофильные соли тераалкиламмония (ТБАБ, ТОАБ, ГДТМАБ) и третичные спирты (2‑метилпропанол-2, 2-метилбутанол-2), проявляют активность на второй стадии дегидробромирования 1,2-дибром-1-фенилэтана, приводящей к образованию фенилацетилена.
Практическая значимость работы. Продукты реакций элиминирования являются ценным сырьем для получения различных органических соединений. В работе предложены эффективные межфазные каталитические системы для селективного синтеза целевых продуктов дегидробромирования 1,2-дибром-1-фенилэтана — a-бромстирола и фенилацетилена — с высоким выходом при практически полном отсутствии продуктов нуклеофильного замещения, характерных для процессов в гомогенных системах. Использование смесей катализаторов межфазного переноса и сокатализаторов кислотно-основной природы, обладающих синергизмом действия, позволяет расширить круг субстратов для получения алкенов и алкинов в МФК/ОН--системах, преимущества которых заключаются в простоте аппаратурного оформления, минимальных расходах органических растворителей и реагентов, мягких условиях проведения реакций.
Гранты. Работа поддержана грантами: а) федеральной целевой программы «Интеграция науки и высшего образования России на годы» (З 4348, 2004 г.), б) грантами программы «Поддержка научно-исследовательской работы молодых ученых и аспирантов ТюмГУ» (2005 г., 2006 г.).
I. ЛИТЕРАТУРНЫЙ ОБЗОР
Реакции элиминирования в условиях межфазного катализа
1. Современные представления о методе межфазного катализа
Метод проведения реакций в двухфазных системах с использованием межфазных переносчиков и соответствующий термин «межфазный катализ» (МФК) получили широкое признание после публикации Старкса [1]. В настоящее время существует ряд монографий и обзоров [2-19], где подробно расматриваются различные вопросы, связанные с межфазным катализом и проведением реакций в условиях МФК.
Межфазным катализом называют ускорение реакций между химическими соединениями, находящимися в разных фазах. Как правило, это реакции между солями, растворенными в воде или находящимися в твердом состоянии, с одной стороны, и веществами, растворенными в органической фазе, с другой. Катализаторами, ускоряющими массообмен между фазами (межфазный перенос), являются ониевые катионы (Q+) и нейтральные комплексанты неорганических катионов, обладающие липофильностью [2].
Круг реакций, которые могут быть осуществлены в условиях МФК, широк и многообразен. Типичные примеры представлены в табл. 1.1.
Таблица 1.1.
Возможности применения МФК в присутствии гидроксид-ионов (МФК/ОН-) и других ионов (МФК/Х-) [11].
МФК/ОН- | МФК/Х- |
Алкилирование (С-, N-, O-) Карбоприсоединение Элиминирование Конденсация (Виттига, альдольная, Дарзенса) Присоединение по Михаэлю Гидролиз Дейтерообмен Изомеризация Металлорганический синтез | Галогенообмен Этерификация Нуклеофильное замещение (СN-, RS-, N3-, SCN-) Окисление Восстановление Бензоиновая конденсация |
По сравнению с обычными методиками синтеза межфазный катализ [2-4]:
ü не требует дорогостоящих безводных или апротонных растворителей;
ü обеспечивает более высокие скорости реакций при более низких температурах;
ü во многих случаях можно использовать более простое оборудование;
ü вместо дорогостоящих оснований можно использовать водные растворы гидроксидов щелочных металлов.
Кроме того, имеются еще особые преимущества, например: возможность осуществления реакций, которые не идут в других условиях; изменение селективности; изменение соотношения продуктов реакции; более высокие выходы в результате подавления побочных реакций.
Для разработки эффективных межфазных каталитических систем для каждого типа реакций важное значение имеет подбор типа гетерофазной системы, органического растворителя для субстрата, межфазного катализатора (катализатора межфазного переноса), интенсивности перемешивания для обеспечения кинетического режима процесса, а также ряда других условий (температуры, соотношения концентраций реагентов и т. д.).
1.1 Типы гетерофазных систем.
Метод межфазного катализа можно использовать в различных гетерофазных системах: "жидкость – жидкость", "жидкость – твердая фаза", "жидкость – твердая фаза – жидкость" [5].
Система ²жидкость/жидкость². В качестве органической фазы используется неполярный, нерастворяющий воду растворитель. Как правило, это углеводороды (толуол, бензол и др.) или их хлорпроизводные (хлороформ, хлористый метилен, дихлорэтан, хлорбензол), иногда в качестве органической фазы выступает сам субстрат. В качестве водной фазы используются нейтральные или кислые растворы солей либо концентрированные щелочные растворы (50 %-ный КОН и т. п.). Эти два типа водных фаз образуют два типа систем ж/ж, которые отличаются механизмом МФК: в первой системе стадия образования продукта протекает в органической фазе, во второй приобретают значение стадии, связанные с ПРФ [5].
Как известно из литературных данных [2], в системе ж/ж часто происходит соэкстракция некоторого количества гидратной воды, которая может мешать протеканию реакции. Во избежание этого эффекта используют систему ж/т.
Система ²жидкость/твердое тело². Здесь в качестве органической фазы используют те же самые растворители. По типу твердой фазы эти системы можно разделить на три группы. В первой группе твердой фазой являются порошкообразные или таблетированные щелочи или кристаллические измельченные карбонаты, гидриды и другие соединения, способные депротонировать субстрат НА на поверхности раздела фаз и тем сам генерировать анион А , который участвует в дальнейшей реакции. Катализатор Q+ переносит его с поверхности твердой фазы в органический слой в форме ионной пары Q
, который участвует в дальнейшей реакции. Катализатор Q+ переносит его с поверхности твердой фазы в органический слой в форме ионной пары Q X. Во второй группе твердой фазой являются ионофорные соли МY, анионы которых Y и являются одним из реактантов. Катализатор в этом случае способствует разрушению решетки соли МY и солюбилизации аниона Y в органической фазе. С этой целью кроме Q часто используют краун-эфиры и криптаты. В третьей группе в качестве твердой фазы выступает катализатор: например, это может быть обычная или специально приготовленная анионообменная смола. В органической фазе находится субстрат, который реагирует с анионом, находящимся на поверхности смолы. Этот хорошо известный процесс и его варианты следует также рассматривать как МФК [5].
X. Во второй группе твердой фазой являются ионофорные соли МY, анионы которых Y и являются одним из реактантов. Катализатор в этом случае способствует разрушению решетки соли МY и солюбилизации аниона Y в органической фазе. С этой целью кроме Q часто используют краун-эфиры и криптаты. В третьей группе в качестве твердой фазы выступает катализатор: например, это может быть обычная или специально приготовленная анионообменная смола. В органической фазе находится субстрат, который реагирует с анионом, находящимся на поверхности смолы. Этот хорошо известный процесс и его варианты следует также рассматривать как МФК [5].
Система ²жидкость/жидкость/твердое тело². Это так называемый трехфазный катализ. Две жидкие фазы — это обычная для МФК система ж/ж, а твердая фаза — это полимер краун-эфира или какой-либо носитель с "пришитыми" к нему четвертичными аммониевыми ионами или краун-эфирами. Достоинством данной системы является то, что твердый катализатор легко отделяется от реакционной массы.
Кроме того, существует ряд работ [20-24], в которых описывается возможность генерирования при определенных условиях трехфазной системы в присутствие аммониевых солей различного строения.
Успешно могут работать и системы ж/т/т, в которых и катализатор и один из реагентов находится в твердом виде, однако такие системы мало изучены [5].
1.2 Межфазные катализаторы и механизм их действия.
Наиболее часто в качестве катализаторов межфазного переноса анионов используют липофильные катионы Q+ ониевых солей или комплексообразователи, которые могут связывать катионы щелочных металлов с образованием липофильных катионов.
Заряженные катализаторы: четвертичные ионы. Первыми катализаторами, используемыми в качестве межфазных, были четвертичные ониевые соли [7]. В настоящее время в качестве катализаторов межфазного переноса предлагается много четвертичных аммониевых, фосфониевых, арсониевых, висмутониевых и третичных сульфониевых солей [2, 7, 8, 25-31].
Первоначальный механизм МФК, предложенный Старксом для реакций замещения, приведен ниже [2]:
Схема 1.1
Механизм МФК для реакций нуклеофильного замещения по Старксу
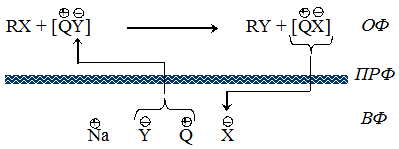
Ионная пара Q+Y-, возникающая при экстракции аниона Y![]() в органической фазу с помощью ониевого катиона Q+, быстро вступает в реакцию обмена с RX. Образующаяся при этом новая соль Q+X - возвращается в водную фазу, где Q+ снова захватывает ион Y
в органической фазу с помощью ониевого катиона Q+, быстро вступает в реакцию обмена с RX. Образующаяся при этом новая соль Q+X - возвращается в водную фазу, где Q+ снова захватывает ион Y![]() и начинается следующий цикл [8].
и начинается следующий цикл [8].
Независимо друг от друга Брендстрем и Монтанари показали, что для проявления МФК в этих реакциях нет необходимости в миграции катиона катализатора в водную фазу. Для реальных процессов МФК с применением липофильных катализаторов предложена исправленная схема Старкса [2]:
Схема 1.2
Исправленная схема Старкса

Катализатор в реакциях, где нуклеофильный анион уже имеется в одной фазе (водной) и необходимо только перевести его в другую (органическую), должен [6]:
– переносить один реагент в ту фазу, где находится другой реагент;
– не связываться с перенесенной частицей слишком прочно, т. е. она должна находиться в окружении, подходящем для реакции;
– не участвовать в реакции в качестве одного из реагентов.
Эффективность ониевых катализаторов на примере реакции тиофенолят-иона c н-бромоктаном
![]() (1.1)
(1.1)
детально изучили Херриот и Пиккер [32]. На основании полученных в этой работе данных, авторы сделали несколько обобщений, которые применимы и для других гетерогенных систем, если реакция преимущественно протекает в органической фазе [5]:
1. Большие четвертичные ионы эффективнее ионов меньшего размера.
2. Каталитическая активность повышается с увеличением размера наиболее длинной цепи.
3. Более симметричные ионы гораздо эффективнее, чем ионы только с одной длинной цепью.
4. Ионы фосфония несколько более эффективны и термически более устойчивы, чем соответствующие аммониевые катализаторы, а фосфониевые и аммониевые катализаторы эффективнее, чем арсониевые системы.
5. Лучшими катализаторами являются четвертичные ионы с алкильными, а не с арильными заместителями.
Следует отметить, что эффект МФК для огромного количества реакций субстратов с рядом анионов (галогениды, циониды, тиоционаты и т. д.) можно описать моделью экстракционного механизма, т. е. переносом анионов из водной или твердой фазы с помощью липофильного катиона межфазного переносчика в объем органической фазы. Однако классические межфазные переносчики не способны эффективно переносить в органическую фазу высоко гидрофильные анионы, такие как ОН–. В этом случае реализуется механизм с участием поверхности раздела фаз (ПРФ), который впервые был предложен Макошей [12] для МФК/ОН–-систем. Для реакций алкилирования Макоша [12] предложил следующую схему механизма (схема 1.3).
Схема 1.3
Схема механизма на ПРФ

Согласно межфазному механизму, молекула субстрата RH депротонируется ОН-–ионом на ПРФ. Если катализатор в системе отсутствует, то на поверхности раздела фаз образуется как бы двухслойная структура, включающая со стороны водной фазы катион щелочного металла, а со стороны органической фазы депротонированный анион субстрата. Из-за взаимной нерастворимости в противоположных фазах ионы иммобилизуются и в значительной степени дезактивируются. Эта ситуация похожа на обычную адсорбцию на поверхности. Катион катализатора в таких случаях способствует транспорту карбаниона R - вглубь органический фазы, одновременно происходит переход Х - в водную фазу. И, наконец, ионная пара Q+R - реагирует с алкилирующим агентом RY с образованием R-R, а образовавшаяся ионная пара Q+Y - включается в каталитический цикл [2, 11].
Этот механизм доказан для реакций С‑алкилирования [33-39]. В частности, в результате детальных исследований реакции алкилирования малонового эфира аллилхлоридом [39] установлено, что при реализации механизма реакции алкилирования с участием ПРФ ключевыми факторами, определяющими эффективность действия межфазных катализаторов, является их способность смещать равновесие депротонирования субстрата на ПРФ вправо за счет стабилизации аниона субстрата в составе ионной пары Q+Sub–, а также способность переносить карбанион Sub– с ПРФ в органическую фазу. Показано, что активность длинноцепочечных солей алкилтриметиламмония возрастает с увеличением длины углеводородного радикала: С12 < С14 < С16. Это может быть обусловлено тем, что с увеличением липофильности катиона межфазного переносчика Q+ возрастает его способность к переносу в органическую фазу карбаниона субстрата в составе ионных пар Q+Sub–, т. е. ряд активности согласуется с закономерностями, характерными для экстракционного механизма. Однако при этом отмечается, что в отличие от экстракционного механизма симметричные катионы (R)4N+ менее эффективны, чем катионы с одной длинной цепью R(CH3)3N+. Авторами работы [40] высказано предположение, что для реакций, протекающих с участием ПРФ, каталитическая активность четвертичных алкиламмониевых солей (ЧАС) симметричного строения коррелирует с их способностью снижать межфазное натяжение на границе раздела водной и углеводородной фаз в системах «ж/ж». Авторы считают, что в таких системах поверхностная активность ЧАС является доминирующим фактором при выборе катализатора межфазного переноса.
К числу реакций, протекающих только на ПРФ в МФК/ОН–-системах относятся некоторые реакции элиминирования. В [41] для реакции дегидробромирования 1,2-дибром-1-фенилэтана в системах «ж/ж» и «ж/т» предложен следующий механизм (схема 1.4).
Схема 1.4
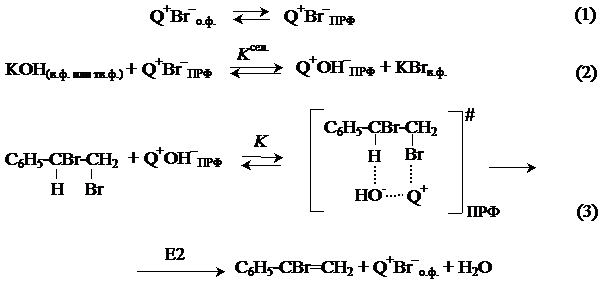
Согласно этой схеме межфазный катализатор распределяется между органической фазой и ПРФ (1), так как из концентрированного водного раствора щелочи он практически полностью высаливается. На ПРФ осуществляется ионный обмен с образованием липофильной ионной пары Q+OH‾ (2), которая затем вступает во взаимодействие с субстратом (3). Предполагается, что стадия химического взаимодействия протекает на ПРФ.
Показано, что в этой реакции соли тетраалкиламмония обладают достаточно высокой каталитической активностью как в системах «ж/т», так и в системах «ж/ж», причем в ряду гомологов: (CH3)4NBr, C14H29(CH3)3NBr, C16H33(CH3)3NBr активность возрастает с увеличением размера длинноцепочечного радикала, т. е. с увеличением липофильности. При этом активность соли (C4H9)4NBr с катионом симметричного строения выше активности солей C14H29(CH3)3NBr и C16H33(CH3)3NBr с длинноцепочечным катионом в системах «ж/т» и ниже в системах «ж/ж».
Авторами работы [41] высказано предположение, что на эффективность межфазного переноса может влиять доступность положительного заряда на атоме азота в катионе тетраалкиламмония для анионов разного размера в зависимости от размера алкильных заместителей. Для анионов малого размера и, в частности, для негидратированных ионов ОН– в системах «ж/т», доступность заряда на атоме азота не зависит существенно от размера алкильных заместителей, и ряд активности солей согласуется с увеличением их липофильности. Для объемных гидратированных ионов ОН– в системах «ж/ж» доступность заряда на атоме азота существенно зависит от размера алкильных заместителей. Образование «рыхлых» ионных пар с катионами тетраалкиламмония симметричного строения приводит к снижению эффективности межфазного переноса
Незаряженные катализаторы: краун-эфиры. МФК с использованием краун-эфиров несколько отличается от катализа с участием четвертичных ионов. Краун-эфиры в основном проявляют активность в системах «жидкость/твердое тело» [8, 42].
Успех катализируемых краун-эфирами межфазных процессов зависит от нескольких факторов. Во-первых, краун-эфир должен быть способен к комплексообразованию с твердым реагентом. Во-вторых, комплекс краун-эфира с солью должен растворяться в реакционной среде. В результате, при прочих равных условиях, чем более липофильными будут краун-эфиры, тем более эффективными катализаторами они окажутся в процессах межфазного переноса. Наконец, между комплексом "металл – краун-эфир", нуклеофилом и нуклеофугом должно существовать равновесие, обеспечивающее легкий обмен анионами [8].
M+Nu–(т. ф.) + краун(раствор) ↔ краун M+Nu–(раствор) (1.2)
краун M+Nu– + RY → краун M+Y–(раствор)+ R Nu (1.3)
краун M+Y–(раствор) ↔ краун(раствор) + M+Y–(т. ф.) (1.4)
Молекулярные катализаторы: полиэтиленгликоли. Полиэтилен-гликоли (ПЭГ) являются ациклическими аналогами краун-эфиров. В принципе такие соединения способны катализировать межфазные реакции, но с меньшей эффективностью, чем циклические [7].
Вследствие конформационной гибкости молекул нециклические полиэфиры не могут проявлять селективность в отношении размера катиона. Длинная цепь олигоэтиленгликолей может связывать более одного иона металла. Кроме того, простые полиэфиры содержат концевые группы - СН2-ОН, которые легко подвергаются элиминированию. Однако недостатки ПЭГ как межфазных катализаторов с лихвой возмещаются их дешевизной [7].
В работе [39] изучена активность ПЭГ, олигоэтиленгликолей и их эфиров в межфазной реакции алкилирования малонового эфира аллилхлоридом. Показано, что в системах «ж/ж» их активность очень мала из-за высокой растворимости в щелочной водной фазе. В системах «ж/т» их активность близка к активности краун-эфиров. Активность эфиров олигоэтиленгликолей повышается с увеличением их липофильности, которая достигается за счет снижения степени олигомеризации или за счет увеличения длины алкильного радикала в эфирной группе, причем введение в молекулу катализатора алкильного радикала более эффективно, чем ароматического.
В работах [6, 42-49] было показано, что ПЭГ являются высокоэффективными и селективными катализаторами в реакциях элиминирования в двухфазных системах. В довольно мягких условиях удается получить ряд ацетиленовых и виниловых производных. Любопытно, что при замещении в ПЭГ одной гидроксильной группы алкоксильной каталитическая активность снижается, а замена двух концевых гироксильных групп приводит к полной дезактивации ПЭГ. Эти и другие данные дают основание предполагать, что в этом процессе каталитическим действием обладает основание, имеющее краун-подобную структуру [50].
При изучении реакции дегидробромирования 1,2-дибром-1-фенилэтана [41] показано, что ПЭГ мало эффективен в системах «ж/ж» и высоко эффективен в системах «ж/т». Высказано предположение, что в системах «ж/т», где активность воды крайне низка, ПЭГ способен не только взаимодействовать с катионом щелочи с образованием липофильного катиона и соответствующей ионной пары Q+OH–, но и выступать в качестве основания, т. е. принимать участие в переносе протона от субстрата в органической фазе к гидроксид-иону на ПРФ.
"Трехфазные" катализаторы. Катализаторы, нанесенные на носитель, называют "трехфазными", так как в межфазных реакциях они представляют собой третью нерастворимую фазу. Такими катализаторами могут быть четвертичные ониевые соли [51-54], краун-эфиры [55, 56], полиэфиры [57, 58] и криптаты [55], связанные с полистирольной матрицей. Иногда для МФК–реакций используются технические анионообменные смолы [59]. Каталитическая активность полимерных катализаторов может быть столь же велика, что и у растворимых межфазных катализаторов. Так, некоторые циклические эфиры, иммобилизованные на сшитых полимерах проявляют каталитическую активность, сравнимую с активностью четвертичных аммониевых и фосфониевых солей [55]. В качестве межфазных катализаторов на носителях можно использовать полиэфиры, химически связанные с кремнеземом кремния [7], однако, кремнезем (и оксид алюминия) сам по себе может служить межфазным катализатором, но механизм его действия отличен от случая классического МФК [7, 60]. Появились публикации об использовании новых типов катализаторов межфазного переноса: фталоцианинов [61], триизопропилсиланов [62], каликсаренов [63] и С2-симметричных оптически активных пирролидиновых солей, используемых в качестве хиральных катализаторов межфазного переноса [64, 65].
Сокатализаторы. В последнее время в литературе все чаще встречаются сообщения о применении в межфазно-каталитических реакциях смесей катализаторов межфазного переноса, в частности, каталитической пары "краун-эфир ¾ четвертичная ониевая соль" [66-72], либо "полиэтиленгликоль ¾ четвертичная ониевая соль" [73-75]. Исследование закономерностей в этой области межфазного катализа представляет большой интерес как с точки зрения установления или конкретизации механизмов межфазных реакций, так и с точки зрения поиска или выбора оптимальных межфазно-каталитических систем.
На данный момент в межфазном катализе нет обоснованной классификации синергетических эффектов. Синергизм достоверно установлен только в работе [67], в которой показано, что использование смеси хлорида тетрабутиламмония и триэтиламина приводит к сверхаддитивному выходу дихлорноркарана, образующегося из циклогексана и хлороформа. Для количественной характеристики эффекта синергизма предложена величина kS, числено равная отношению выхода продукта реакции в присутствии смеси катализаторов (h3) к сумме выходов продукта в присутствии индивидуальных катализаторов (h1 + h2) [67]:

Если kS > 1, имеет место синергизм, если kS < 1 ¾ антагонизм и если kS = 1 — сокатализ.
В более точной формулировке в качестве количественной характеристики эффекта предлагается величина S, численно равная отношению приращения константы скорости Dk в присутствии смеси катализаторов к сумме каталитических констант скорости k1 и k2 в присутствии индивидуальных катализаторов [67]:
S = Dk / kad (1.6)
где kad = k1 + k2 (1.7)
Dk = k – kad (1.8)
В работе [70] показан высокий синергический эффект в реакции п‑нитрофенилацетата с твердым гидроксидом натрия в толуоле при использовании в качестве катализатора смеси краун-эфира и четвертичной аммониевой соли. Высказывая гипотезу о механизме синергизма, авторы полагают, что реакция протекает в пограничном слое — т. н. «омега»-фазе, представляющей собой тонкую водную пленку на поверхности твердой фазы, в которой (или на поверхности которой) и осуществляется химическая реакция. Синергизм связан, по-видимому, с образованием предреакционного комплекса с участием нуклеофила, субстрата и обоих катализаторов в «омега»-фазе. Роль краун-эфира в данном процессе может сводиться к разрыхлению кристаллической решетки твердого гидроксида натрия за счет образования комплекса "ОН‾ • Na+ • краун-эфир". Роль аммониевого катиона состоит в образовании ассоциата с гидроксид-ионом и координации его с молекулой субтрата за счет гидрофобных взаимодействий между углеводородными группами катиона соли и неполярными группами субстрата.

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 10 11 |
