
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Можно ли распознать гомологичность, родственность последовательностей, если их сходство лежит ниже уровня в 30%, — т. е. в "сумеречной зоне" или даже ниже ее? Можно — но для этого надо сравнивать интересующую нас последовательность со многими последовательностями семейства, и обращать внимание преимущественно на те позиции в цепи, что доказали свою консервативность именно в этом семействе.
Рисунок 19-2 показывает, что рибонуклеаза Н вируса иммунодефицита человека (HIV) имеет не очень высокое — "на уровне шума" — сходство с другими рибонуклеазами Н, если рассматривать всю цепь (так, из 60 выровненных остатков у нее есть всего 9 общих — 15% совпадений — с рибонуклеазой Н из RSV). Однако это сходство проявляется именно в тех ключевых районах, где все остальные рибонуклеазы похожи друг на друга. Это резко повышает достоверность такого сходства. А если еще учесть, что эти "ключевые районы" совпадений охватывают все аминокислотные остатки активного центра, и что сходство концентрируется в участках вторичной структуры, и что оно охватывает около 30% (а не 15%) остатков гидрофобных ядер белка, — высокая достоверность переходит в уверенность в правильном опознании гомологии.
Для опознания гомологии "новых" последовательностей также удобно пользоваться "консенсусными последовательностями" (Рис.19-4), выведенными для уже изученных белковых семейств и подчеркивающими их наиболее консервативные черты. Иногда такие консенсусные последовательности (снабженные данными по частотам встречаемости остатков в каждом месте цепи) называют "профилями первичных структур".
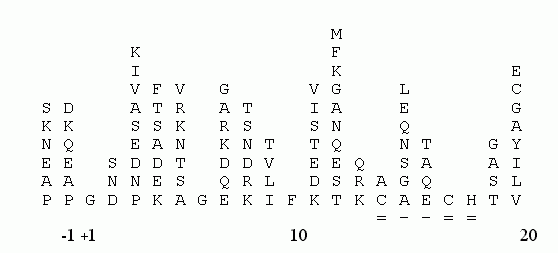
Рис.19-4. Аминокислотный состав различных позиций в N-концевых фрагментах цитохромов c митохондрий эукариот. Самые важные, консервативные остатки цепи определены однозначно. Подчеркнута последовательность "сайта" Cys-X-X-Cys-His, отвечающего за связывание гема в подавляющем большинстве цитохромов (и не только c, и не только эукариот). Выравнивание аминокислотных последовательностей взято из [6].
При опознавании функционального сходства белков следует также обращать внимание на уже установленные для многих функций "сайты" — более или менее короткие последовательности, обеспечивающие эти функции (см. на Рис.19-4 сайт Cys-X-X-Cys-His, связывающий гем в цитохромах). Такие сайты собраны в библиотеки, и их поиском занимаются специальные программы, из которых PROSITE является наиболее популярной.
При установлении структуры "нового" белка по его гомологии с уже изученным надо ясно отдавать себе отчет, что сходство пространственных структур может не распространяться на районы, где последовательности сильно разошлись. В основном это (см. Рис.19-2) районы петель, нерегулярных конформаций белковой цепи. Здесь, с весьма переменным пока успехом, приходится прибегать к конформационным расчетам и другим методам гомологического моделирования, на которых я останавливаться не буду.
Перейдем к методам предсказания пространственной структуры "новых" последовательностей, не имеющих видимой гомологии с уже расшифрованными белками.
К ним относится около 2/3 "новых" последовательностей. Поэтому о пространственной укладке большинства последовательностей, получаемых в ходе выполнения генетических проектов, мы не можем догадаться по их гомологии с белками уже известными — она или слишком слаба для обнаружения, или отсутствует. Тут-то и возникает настоятельная потребность в решении задачи предсказания пространственной структуры — а, в перспективе, и функции белка, — по его аминокислотной последовательности (Рис.19-5).
|
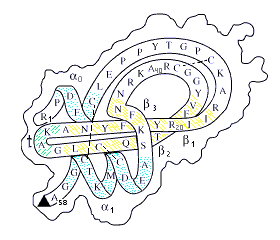
Рис.19-5. Схема первичной и пространственной структуры маленького белка (панкреатического ингибитора трипсина). Ход главной цепи изображен на фоне общего контура молекулы; выделены a-спирали, b-тяжи, резкий поворот цепи (t) и цистеиновые мостики Так как белок сворачивается сам собой, то — в принципе — все это можно предсказать по одной лишь первичной структуре белка. Боковые группы здесь не показаны, но — в принципе — и их расположение в пространстве тоже можно предсказывать.
Надо сразу сказать, что абсолютно надежных и точных методов предсказания белковых структур сейчас нет.
Причин тому, видимо, две: (а) ограниченная точность энергетических оценок, на которых базируется теоретический расчет белковых структур, и (б) сравнительно малая разность между "правильно" и "неверно" уложенными белковыми цепями. Я хочу особо подчеркнуть последнее обстоятельство, — оно резко отличает ситуацию в белках (и РНК) от ситуации в ДНК, — к большому огорчению людей, занявшихся предсказаниями структур белков по их аминокислотным последовательностям. В ДНК комплементарное спаривание нуклеотидов наблюдается по всей длине двойной спирали, а оно обеспечивается тем, что одна цепь по своей первичной структуре строго комплементарна другой. Это и дает огромный свободно-энергетический выигрыш "правильной" структуры ДНК над всеми "неправильными". А в белках (и РНК) ни какой-либо строгой комплементарности, ни следующего из нее огромного энергетического преимущества "правильной" укладки цепи не наблюдается...
Однако существующие методы дают все более полную и надежную информацию о возможном строении (или, чаще, о возможных вариантах строения) белка или, особенно, его структурных элементов.
В прагматическом аспекте, вопрос о предсказании трехмерного строения белка сейчас ставится следующим образом: похожа ли пространственная структура рассматриваемой белковой последовательности на какую-либо из уже известных пространственных белковых структур? Если да, то как рассматриваемая последовательность вписывается в эту структуру? Если нет, можем ли мы указать какие-либо характерные детали пространственной организации рассматриваемой последовательности?
Я знаю несколько случаев, когда ответы на эти вопросы существенно способствовали экспериментальному определению структуры и/или функции изучаемой аминокислотной последовательности, помогли планировать белково-инженерные эксперименты и так далее. Впрочем, должен сказать, что я бы с большим интересом прочел бы хороший обзор о практическом применении предсказаний белковых структур...
Перейдем теперь к методам предсказания белковых структур. Рассматривая эти методы, я буду уделять особое внимание тем идеям, и в основном физическим идеям, что лежат в их основе.
Есть две стратегии предсказания белковых структур. Согласно первой стратегии, белковая структура ищется как результат кинетического процесса сворачивания.
Согласно второй, — она ищется как структура с минимально возможной для данной цепи свободной энергией.
В принципе, по-видимому, обе эти стратегии могут привести к правильному результату, так как самая стабильная структура белковой цепи, несмотря на опасения Левинталя, образуется достаточно быстро (этому вопросу была посвящена отдельная лекция, и сейчас я позволю себе его не касаться).
Важно, однако, что, рассматривая предсказание белковых структур, мы можем отвлечься от путей сворачивания и рассматривать только результат — стабильную структуру белка. Тем более, что первая стратегия — стратегия имитационного моделирования процесса сворачивания белка — пока существенного прогресса не дала: уж очень сложны все расчеты, и к тому же — все же весьма приблизительны все потенциалы взаимодействий. Вторая стратегия оказалась более успешной.
Начнем с определения стабильной вторичной структуры белка по его аминокислотной последовательности. a-спирали и вытянутые b-участки — важнейшие элементы белковых глобул, во многом определяющие, как вы помните, их общую архитектуру (Рис.19-5).
Забудем пока о том, что белковая цепь свернута в глобулу, т. е. рассмотрим развернутую белковую цепь. Можем ли мы предсказать ее вторичную структуру по аминокислотной последовательности? Оказывается — да, в основном да, пусть и не абсолютно точно.
Прежде всего, — какие аминокислотные остатки будут стабилизировать вторичную структуру — например, a-спираль, — а какие будут разрушать ее?
Эксперименты дают прямой ответ на этот вопрос. Я имею в виду огромную работу по оценке a - и b-образующих способностей аминокислотных остатков, проведенную в группах Шераги, Фасмана, Болдвина, Фершта, Серрано, ДеГрадо, Кима и в ряде других групп (и, в частности, у нас — и ). Кроме того, богатый (и хорошо совпадающий с физико-химическим экспериментом) материал дает статистика аминокислотных последовательностей a - и b-структур в белках. Рисунок 19-6 суммирует важнейшие (те, которые стоит запомнить) из оценок, полученных всеми этими методами.
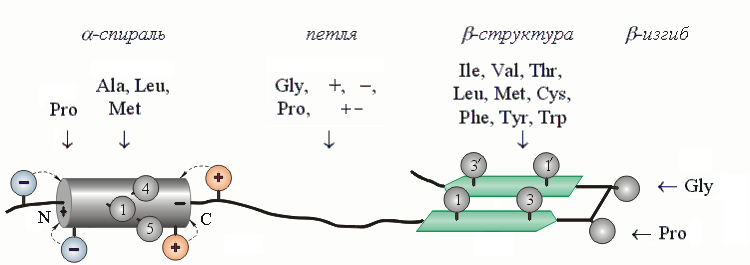
Рис.19-6. Шаблоны a-спирали, петли, b-структуры и b-изгиба — те остатки, которые стабилизируют их или их отдельные части. "+" означает все положительно заряженные аминокислоты, "-" — все отрицательно заряженные, "+ -" — все аминокислоты с диполем в боковой цепи. Показан также стабилизирующий a - и b-структуру порядок чередования гидрофобных групп в цепи (см. нумерованные группы). Такого типа чередование приводит также к образованию гидрофобных и полярных поверхностей a-спиралей и b-тяжей.
Здесь надо, однако, сразу сказать, что все закономерности, наблюдающиеся в структурах белков, носят вероятностный, частотный характер. Несмотря на многочисленные попытки, не удалось выделить никакого четкого "кода" белковых структур — то есть ничего похожего на то четкое A-T и G-C спаривание нуклеотидов, которое свойственно двойной спирали ДНК. Впрочем, надо признать, что уже в РНК — с их разнообразным, в отличие от ДНК, репертуаром пространственных структур — спаривание нуклеотидов происходит далеко не столь однозначно...
Подавляющее большинство из полученных экспериментальных и статистических оценок можно легко понять, исходя из физики и стереохимии аминокислот. Мы уже говорили об этом на одной из прошлых лекций.
Так, Pro не любит входить ни в a-спираль (кроме ее N-концевого витка), ни в b-структуру. Почему? Потому, что у него нет NH-группы, и он не может завязывать соответствующие a - и b-структуре водородные связи; а на N-конце спирали NH-группа таких связей и не должна завязывать, — и Pro там встречается часто, тем более что его угол y уже фиксирован пролиновым кольцом в подходящем положении. По тем же причинам часто встречается Pro и на N-конце изгибов.
А вот Ala стабилизирует a-спираль, — а Gly разрушает и ее, и b-структуру, и способствует образованию клубка. С чем связана эта разница? С тем, что область конформаций, т. е. область допустимых углов f, y для Gly в клубке гораздо больше, чем для Ala, — в то время как допустимые конформации для обоих этих остатков в a-спирали примерно совпадают.
По аналогичным причинам разветвленные остатки — Val, Ile и Thr — больше стабилизируют b-структуру, где их боковые группы имеют 3 разрешенных поворотных изомера, чем a-спираль или клубок, где они имеют только 1 изомер (при каждом значении f и y).
Вообще же гидрофобные группы склонны несколько чаше входить в a - и b-структуру, где они могут слипаться в гидрофобных кластерах (см. Рис.19-6), чем в клубок, где они этого делать не могут. А вот боковые группы с диполями, особенно — в короткой боковой цепи, больше склонны входить в нерегулярные участки цепи, где они могут завязывать дополнительные нерегулярные водородные связи, чем в a - и b-структуры, где доноры и акцепторы водородных связей уже насыщены внутрицепными связями.
Более того, влияние аминокислотных остатков на вторичную структуру можно оценить теоретически. Так, еще до получения экспериментальных оценок, в начале и середине 70-х годов, мы с предсказали, что отрицательно заряженные остатки должны стабилизировать N-конец спирали, притягиваясь к его положительному парциальному заряду, и дестабилизировать ее С-конец, отталкиваясь от отрицательного парциального заряда спирального диполя. Положительно же заряженные остатки должны действовать в прямо противоположном направлении. При этом потенциал каждого такого взаимодействия должен, по теоретической оценке, составлять 1/4 или 1/3 килокалории. Что и было подтверждено экспериментально.
Зная, какие аминокислотные остатки стабилизируют середину спирали, какие — ее N-конец, а какие — C-конец, мы получаем нечто вроде "шаблона" спирали. Шаблон a-спирали можно описать так: если начало фрагмента белковой цепи обогащено отрицательно заряженными группами, да еще там стоит Pro; если середина этого фрагмента обогащена остатками Ala, Leu и Met, а Pro и Gly там нет; и если его С-конец содержит положительно заряженные боковые группы, — то перед нами a-спиральный участок. Кроме того, для стабильности a-спирали полезен, а для ее включения в глобулу — просто необходим определенный (Рис.19-6) порядок чередования гидрофобных групп в цепи: этот порядок способствует слипанию этих групп и, кроме того, приводит к образованию на спирали сплошной гидрофобной поверхности, необходимой для ее прилипания к глобуле.
То есть "шаблон" качественно описывает аминокислотную последовательность, подходящую для образования a-спирали. Чем лучше аминокислотная последовательность удовлетворяет этому шаблону — тем вероятнее спираль в данном месте цепи. Такое же описание — "шаблон" — можно составить и для участков, пригодных для образования b-структуры. А также — для участков, особо пригодных для образования b-изгибов и петель.
Более того, "шаблоны" можно применять и для описания кусков цепи, образующих более сложные структуры, — например, для описания b-a-b суперспиралей, состоящих из двух параллельных b-участков и a-спирали между ними (Рис.19-7). Такие структуры типичны для доменов, связывающих нуклеотиды. Особо важную роль в "шаблоне" играют так называемые "ключевые позиции", которые могут быть заняты только строго определенными аминокислотными остатками, — например, Gly: только этот остаток может находиться в конформации с f>60о, недоступной всем остальным остаткам.
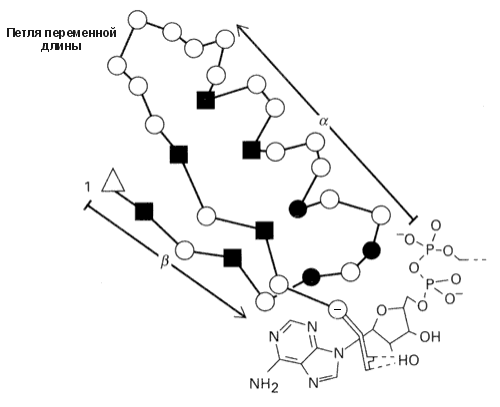
Рис.19-7. "Шаблон" суперспирали b-a-b, связывающей нуклеотиды. Квадратики отмечают ключевые позиции, обычно занимаемые сравнительно небольшими гидрофобными остатками (Ala, Ile, Leu, Val, Met, Cys): это — гидрофобное ядро суперспирали b-a-b. Черные кружки — позиции, занимаемые только Gly: здесь находятся резкие повороты цепи. Пустой треугольник отмечает первую позицию мотива b-a-b, где обычно находится положительно заряженная или дипольная боковая цепь. В последней (-) позиции мотива b-a-b находится аминокислота Asp или Glu, связывающая лиганд (нуклеотид). Рисунок, с небольшими изменениями, взят из R. K.Wierenga et al., J. Mol. Biol. (1986) 187:101-107.
Но вернемся к расчету вторичной структуры. Зная вклады отдельных взаимодействий в стабильность a-спирали, мы можем рассчитать свободную энергию спирализации любого участка цепи, а следовательно — и Больцмановскую вероятность образования спирали в любом месте полипептидной цепи, еще не свернувшейся в глобулу. Суммируя и усредняя эти вероятности, мы можем рассчитать и среднюю спиральность такого "несвернутого" полипептида. Вот уже более 15 лет мы используем для этого нашу программу ALB. В одном из режимов ("unfolded chain" — "развернутая цепь") она позволяет рассчитывать содержания a - и b-структуры в полипептидах и в несвернутых белковых цепях, — причем при разной температуре, ионной силе и рН раствора. Потом результат можно сравнить с опытными данными — например, с КД спектрами. Рисунок 19-8 показывает, что теоретический расчет неплохо совпадает с опытом.
| Рис.19-8. Теоретическая (вычисленная программой ALB — unfolded chain) и экспериментально найденная спиральность нескольких десятков пептидов при температуре 0о-5оС и разной ионной силе и рН раствора. Рисунок взят из A. V.Finkelstein, A. Y.Badretdinov & O. B.Ptitsyn, Proteins (1990) 10:287-299. |

Переходя к расчету и предсказанию вторичной структуры белков, глобулярных белков, необходимо учесть, что здесь к взаимодействиям, существующим в несвернутых цепях, добавляется взаимодействие каждого участка цепи с глобулой, строения которой мы не знаем. Точнее, мы не знаем ее детального строения, но знаем, что участки цепи как-то примыкают к гидрофобному ядру белка. В простейшем приближении взаимодействие с ядром можно аппроксимировать взаимодействием с "гидрофобным озером", на котором плавает белковая цепь (Рис.19-9).
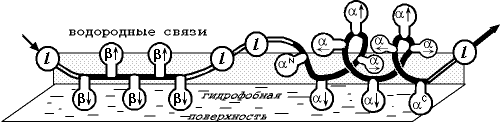
Рис.19-9. Флуктуирующая вторичная структура белковой цепи (здесь: b — b-тяж, l — петля, a — a-спираль) на поверхности гидрофобного озера, имитирующего белковую глобулу ("модель плавающих бревен"). Модель учитывает разное чередование обращенных к поверхности озера (![]() ), от поверхности (
), от поверхности (![]() ) и вдоль нее (®, ) боковых групп в разных вторичных структурах, а также эффекты на N - и С-концах спирали, объединенные и приписанные ее, соответственно, N - и С-концевым остаткам (aN, aC). Рисунок, с небольшими изменениями, взят из O. B.Ptitsyn & A. V.Finkelstein, Bipolymers (1983) 22:15-25.
) и вдоль нее (®, ) боковых групп в разных вторичных структурах, а также эффекты на N - и С-концах спирали, объединенные и приписанные ее, соответственно, N - и С-концевым остаткам (aN, aC). Рисунок, с небольшими изменениями, взят из O. B.Ptitsyn & A. V.Finkelstein, Bipolymers (1983) 22:15-25.
Зная из опыта силу гидрофобных взаимодействий, а из стереохимии a - и b-структуры — мотивы чередования в цепи боковых групп, глядящих в одну и ту же сторону и способных, следовательно, одновременно взаимодействовать с гидрофобной поверхностью, — мы можем сосчитать вероятность образования a-спирали и b-структуры в каждом месте белковой цепи. Этим также — но уже в режиме "глобулярная цепь" ("globular chain") — также занимается программа ALB (кстати, к ней можно обращаться по Интернет: http://indy. ipr. serpukhov. su/~rykunov/alb/).
Вероятности рассчитываются при комнатной температуре. Почему при комнатной? Не лучше ли выделять только самое лучшее по энергии расположение a - и b-структур в цепи, т. е. рассчитывать все вероятности при 0оК?
Прежде всего, конечно, расчет относится к комнатной температуре потому, что все экспериментальные оценки стабильности, на которых базируется расчет, относятся к этой температуре.
Но еще важнее то, что вероятности, рассчитанные именно при такой температуре (300оК), лежащей чуть ниже нормальной температуры плавления белка (350оК) наилучшим образом позволяют отделить более вероятные a - и b-участки, в которых мы можем быть более или менее уверены, от тех "менее вероятных", в которых мы уверенными быть не можем.
Ведь, в сущности, мы стараемся предсказать вторичную структуру белка только по части тех взаимодействий, что ее держат в действительности: мы знаем (и то приблизительно) внутренние взаимодействия в этой структуре, но не знаем (или можем лишь крайне грубо оценить) те, что действуют между рассматриваемым куском цепи и остальной глобулой. А они очень мощны.
То есть мы находимся в том же положении, как если бы пытались предсказать, будет ли данный остаток внутри белка или на его поверхности, зная только его собственную гидрофобность — и больше ничего. И мы знаем, что такая задача имеет ответ, — но ответ не точный, а вероятностный. Этот ответ содержится в наблюдаемой статистике распределения остатков между нутром и поверхностью глобулы. Мы уже знаем, что она выглядит так:
ВЕРОЯТНОСТЬ_ВНУТРИ / ВЕРОЯТНОСТЬ_НА_ПОВЕРХНОСТИ ~ | (19.1) |
где TС — характерная температура белковой статистики, лежащая — как мы тоже помним — несколько ниже температуры плавления белка.
И — возвращаясь к предсказаниям белковых структур — по той же формуле (19.1), и с той же температурой ТС мы можем вероятностно предсказывать существование рассматриваемой вторичной структуры, зная только ее собственную свободную энергию.
Рисунок 19-10 иллюстрирует расчет вторичной структуры в глобуле. Он был сделан в 1985 г. для интерферонов. Видно, что в них преобладают a-спирали, особенно в N-концевой части цепи, где (как было известно из других опытов) находится функциональный домен интерферона. В данном случае спирали эти предсказывались столь уверенно (а это бывает отнюдь не всегда) и столь одинаково в разных, не так уж и высоко гомологичных цепях, что мы попробовали — в том же 1985 г. — слепить домен из этих спиралей.
| Рис.19-10. Расчет вторичной структуры нескольких интерферонов. Абсцисса — номер остатка в белковой цепи, ордината — вычисленная вероятность его a-спирального ( ____ ) и b-структурного ( _ _ _ ) состояния. Сверху приведены предсказания наиболее вероятных a-спиралей (a), b-тяжей (b) и изгибов (Т). Черные прямоугольники — уверенное предсказание вторичной структуры, пустые — не столь уверенное предсказание, линии — данная вторичная структура в принципе не исключена, но маловероятна. Цепь, образующая N-концевой домен, подчеркнута внизу рисунка. Картинка взята из O. B.Ptitsyn, A. V.Finkelstein & A. G.Murzin, FEBS Letters (1985) 186:143-148. |
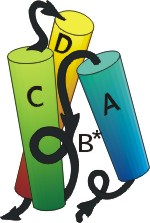
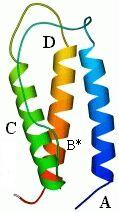
Полученный комплекс, пучок из трех больших спиралей и одной крошечной, показан на следующем рисунке (19-11а). Через пять лет после того, как было сделано это предсказание, была, наконец, получена рентгеновская структура интерферона b (Рис.19-11б), — и рентгеновская структура N-концевого его домена довольно точно совпала с той, что была предсказана нами.
|
|
|
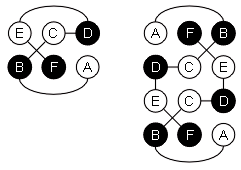
а б в
Рис.19-11. (а) Предсказание укладки цепи в N-концевом домене интерферонов, сделанное нами в 1985 г. [Ptitsyn, Finkelstein, Murzin, FEBS Letters (1985) v.186, p.143]. Три большие a-спирали (А, C, D) изображены цилиндрами; кроме того, была предсказана возможность существования отдельного спирального витка В. (б) Рентгенографическая структура N-концевого домена (С-концевой домен — не изображен) b-интерферона, расшифрованного в 1990 г. [Senda et al., Proc. Jpn. Acad. Sci., ser. B, Biopys. Biol. Sci. (1990) v.66, p.77]. Рисунок (б) дан в той ориентации, что и предсказанная модель (а), и на нем даны те имена больших спиралей (А, C, D), под которыми они значатся на рисунке (а). Район В* спиралеподобен, но не a-спирален в интерфероне b, однако в родственном ему интерфероне g там находится небольшая a-спираль [Ealick et al., Science (1991) v.252, p.698]. (в) Топологии b и g интерферонов по Ealick et al. Интерферон g состоит из двух субъединиц. Обратите внимание на "обмен" С-концевыми доменами [спиралями (E, F)] между этими двумя субъединицами.
Структура N-концевого домена интерферона — одна из первых, более или менее успешно предсказанных до опыта структур белковых молекул.
Хочу подчеркнуть факторы, способствовавшие успеху предсказания: a-спирали предсказаны здесь очень уверенно; и они одинаково расположены в отдаленно-гомологичных цепях, что позволяет им доверять.
Однако столь уверенное и повторяющееся в гомологах предсказание вторичной структуры встречается на практике не часто. Более типичны некоторые разночтения, как в С-концевом домене интерферонов (Рис.19-10), для которого — именно вследствие этого, очень типичного небольшого разночтения — однозначного предсказания укладки цепи получить не удалось.
И еще очень важная вещь для успеха предсказания строения интерферона. Когда мы искали наилучшую упаковку предсказанных a-спиралей, мы могли рационально организовать перебор всех возможных упаковок, так как располагали априорной классификацией a-спиральных комплексов (я об этом уже говорил) — она как раз была разработана к тому времени Мурзиным и мной.
Вернемся к предсказанию вторичной структуры.
Оно носит вероятностный характер: точность распознавания a-, b-структуры и нерегулярных петель белка по его аминокислотной последовательности составляет около 65%.
Можно ли улучшить предсказание вторичной структуры? Да. В методе "PHD" Роста и Сандера (очень его рекомендую, он доступен по Интернет) вторичная структура предсказывается не для одной аминокислотной последовательности, а для набора гомологов. В результате такого подхода случайные погрешности как бы сглаживаются и предсказание становится намного более точным (достигая, в среднем, 72-73% вместо 63-65%).
Итак — несмотря на свою ограниченную точность, предсказание вторичной структуры стало уже, по сути, рутинной процедурой исследования первичной структуры белка.
Перейдем теперь к новой, быстро развивающейся области — к работам по предсказанию пространственных укладок белковых цепей.
Проблему предсказания белковой структуры можно представить себе как проблему выбора той структуры, которая наиболее стабильна для данной аминокислотной последовательности. Проблема заключается в том, откуда взять "возможные белковые структуры". Самый простой, прагматичный ответ — взять из Банка Белковых Структур. Там, конечно, нет еще всех возможных структур — но есть надежда, что там есть примерно половина всех существующих в природе мотивов укладки белковых цепей в домены. Эту надежда — ее обосновал Сайрус Чотиа — основана на том, что вновь расшифровываемые белки (точнее — их домены) все чаще и чаще оказываются похожими на уже расшифрованные.
Кстати, сейчас разворачивается большой проект "Structural Genomics", цель которого — расшифровать пространственное строение хотя бы одного представителя каждого семейства белков (а их — порядка 10000). Когда эта программа будет завершена — для этого придется потратить примерно 10 миллиардов $ за 10 лет — все предсказание структур "новых" белков можно будет — мы надеемся на это — делать по гомологии с уже расшифрованными белками. Однако пока что приходится использовать лишь вероятностные методы предсказания; в настоящее время они — точнее, лучшие из них — дают правильный ответ (для белков, не имеющих видимой гомологии с уже расшифрованными) примерно в 30% случаев.
Итак, предсказывая структуру белка, не имеющего видимой гомологии с белками уже расшифрованными, можно попробовать взять, одну за другой, все пространственные структуры из Банка, наложить (возможно, с некоторыми выпетливаниями, см. Рис.19-12) цепь этого белка на каждую из них, и посмотреть, какая из этих пространственных структур даст — для нашей цепи — наибольший энергетический выигрыш. При этом мы должны разрешать цепи то идти по скелету структуры, то выпетливаться или "сокращать" имеющиеся в скелете выпетливания — если это увеличивает энергетический выигрыш.
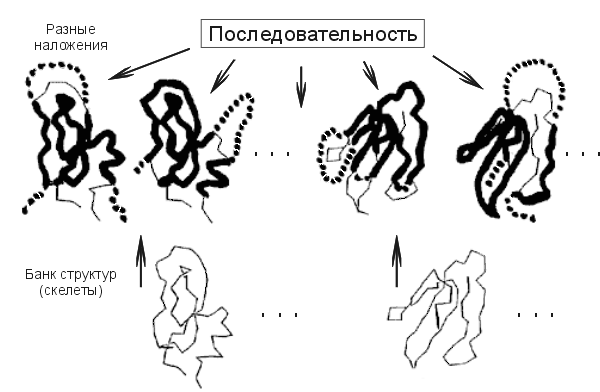
Рис.19-12. Схема, иллюстрирующая идею "протягивания" исследуемой последовательности по Банку Белковых Структур. Жирная линия показывает те участки, где последовательность идет по скелету, пунктир — те, где она выпетливается.
Такой подход называют "методом протягивания" (threading method). Он был предложен нами с в 1990 г. и — независимо, в более простом и более удобном варианте — Д. Айзенбергом и его группой в 1991 г. Сейчас метод протягивания стал весьма популярным методом опознавания структур "новых" белков по их аналогии со "старыми".
В общем, работа по протягиванию напоминает поиск гомологии, — только на этот раз "выравниваются" не две первичные структуры, "новая" и "старая", а "новая" первичная структура со "старым" белковым скелетом.
Прежде, чем перейти к результатам, — хочу подчеркнуть две принципиальные проблемы метода "протягивания". В сущности, аналогичные проблемы, в том или ином виде, возникают в любом "предсказательном" методе.
Во первых, конформацию даже тех кусков цепи, что наложены на скелет, мы знаем с большой погрешностью: ведь мы не знаем конформации боковых групп, — а именно они, в основном, и взаимодействуют. Далее, мы не знаем конформации всех выпетливаний. Оценка показывает, что при протягивании мы знаем примерно половину взаимодействий в белковой цепи, а вторую — не знаем. Значит, опять мы вынуждены судить о структуре белка по части взаимодействий, действующих в его цепи. Значит, опять наши предсказания могут носить только вероятный характер.
Во-вторых, как перебрать все наложения и найти лучшее — или лучшие — среди них? Ведь их, возможных наложений, страшно много... Скажу коротко: мощные математические вычислительные методы для этого уже развиты, но рассказ об этом занял бы слишком много времени. Наверно, имеет все же смысл назвать эти методы по имени — может быть, кому-то из вас пригодится (но большинству, конечно, нет). Итак: динамическое программирование и его вариант — статистическая механика одномерных систем (цепных молекул) — для расчета протягивания цепи через скелет; теория самосогласованного поля — для расчета действующего на цепь молекулярного поля в каждой точке скелета; стохастическая минимизация энергии методом Монте-Карло; а также — разные варианты метода ветвей и границ, и т. д.
Для примера я покажу предсказанную методом протягивания укладку цепи в белке, терминирующем репликацию. Протягивая цепь этого "нового" белка по всем известным трехмерным белковым структурам, Зиппль и его коллеги из Зальцбурга показали, что укладка гистона Н5 наиболее "родственна" этой цепи (Рис.19-13).
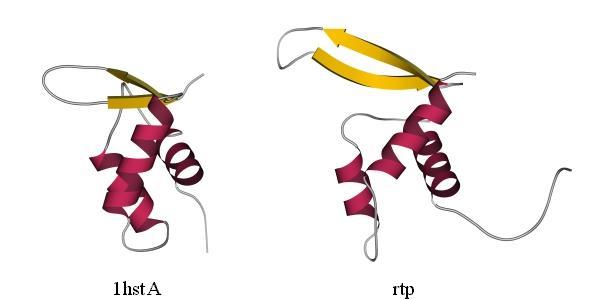
Рис.19-13. Гистон Н5 цыпленка (1hstA, слева) и белок, терминирующий репликацию (rtp, справа); в последнем не показана С-концевая спираль, не имеющая аналога в гистоне Н5. Предсказание сходства укладки цепи в этих двух белках сделано, методом "протягивания", в работе H. Fl![]() ckner, M. Braxenthaler, P. Lackner, M. Jaritz, M. Ortner & M. J.Sippl, Proteins (1995) 23:376-386; это предсказание сделано в ходе "слепого" тестирования предсказательных методов (CASP-1994). Средне-квадратичное отклонение между 65-ю эквивалентными Сa атомами в изображенных структурах — 2.4
ckner, M. Braxenthaler, P. Lackner, M. Jaritz, M. Ortner & M. J.Sippl, Proteins (1995) 23:376-386; это предсказание сделано в ходе "слепого" тестирования предсказательных методов (CASP-1994). Средне-квадратичное отклонение между 65-ю эквивалентными Сa атомами в изображенных структурах — 2.4![]() .
.
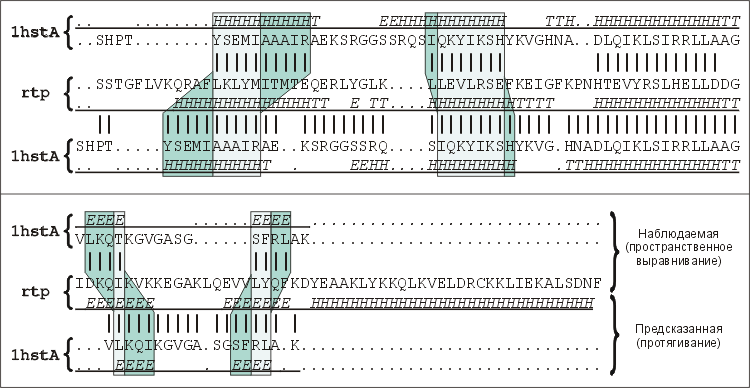
Рис.19-14. Последовательность гистона Н5 (1hstA), выровненная относительно последовательности белка, терминирующего репликацию (rtp) согласно рентгеноструктурным данным ("Наблюдаемая", верхняя пара последовательностей), и она же, выровненная согласно предсказанию методом протягивания ("Предсказанная", нижняя пара). Серыми зонами показаны сдвиги, расхождения этих двух выравниваний. Они хорошо видны по разным сдвигам вторичных структур гистона Н5, приведенных в самой верхней и самой нижней строке в этих двух выравниваниях. Вторичные структуры записаны в подчеркнутых строках (обозначения: Н — a-спираль, Е — b-тяж, Т — изгиб цепи). Точками отмечены делеции, пропуски в первичных и вторичных структурах, внесенные при их выравнивании. Картинка взята из статьи C. M.-R. Lemer, V. J.Rooman & S. J.Wodak, Proteins (1995) 23:337-355, в которой обсуждаются итоги CASP-1994.

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 |
