
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
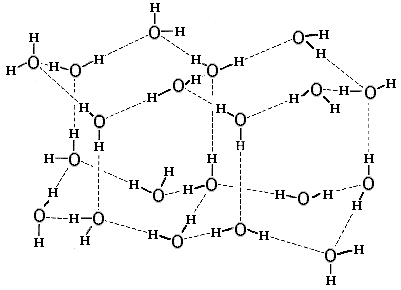
Рис.4-1. Нормальный лед. Пунктир — Н-связи. В ажурной структуре льда видны небольшие полости, окруженные молекулами Н2О. Картинка взята из [6].
Структура нормального льда диктуется Н-связями (Рис.4-1): она хороша для геометрии этих связей (О-Н смотрит прямо на О), но не очень хороша для плотного Вандерваальсового контакта молекул Н2О. Поэтому структура льда ажурна, в нем молекулы Н2О обволакивают микроскопические (размером меньше молекулы Н2О) поры. Ажурность структуры льда приводит к двум хорошо известным эффектам: (1) лед менее плотен, чем вода, он плавает в ней; и (2) под сильным давлением — например, лезвия конька — лед подплавляется.
Большинство из существующих во льду (Рис.4-1) водородных связей сохраняется и в жидкой воде. Это следует из малости теплоты плавления льда (80 кал/г) по сравнению с теплотой кипения воды (600 кал/г при 0оС). Можно было бы сказать, что в жидкой воде рвется только 80/(600+80) = 12% из существующих во льду Н-связей. Однако эта картина — что часть водородных связей в воде разорвана, а часть сохранена — неточна: скорее, все водородные связи в воде разбалтываются. Это хорошо иллюстрируется следующими экспериментальными данными.
На приведенном внизу Рисунке 4-2, кривая 1 соответствует максимуму инфракрасного спектра поглощения групп О-Н во льду (где все Н-связи завязаны); кривая 2 соответствует максимуму инфракрасного спектра поглощения групп О-Н отдельных молекул Н2О, растворенных в CCl4 (где Н-связей нет — раствор Н2О в CCl4 слишком разбавлен); а кривая 3 соответствует спектру поглощения жидкой воды. Если бы в жидкой воде было бы два сорта О-Н групп — образующие водородные связи и не образующие их — и одни О-Н группы в воде колебались бы так же (с той же частотой), как во льду (где они образуют Н-связи), а другие — как в окружении CCl4 (где они Н-связей не образуют). Тогда спектр воды имел бы два максимума, соответствующие двум состояниям О-Н групп, двум их характерным частотам колебаний: с какой частотой группа колеблется, с такой и поглощает свет. Но "двухмаксимумная" картина не наблюдается! Вместо нее на кривой 3 мы видим один, очень размытый максимум, простирающийся от максимума кривой 1 до максимума кривой 2. Это значит, что все О-Н группы в жидкой воде завязывают водородные связи — но все эти связи разболтаны, причем по-разному.
Это показывает, что картина, в которой часть водородных связей в воде разорвана, а часть сохранена, - строго говоря, неверна. Однако она столь проста и удобна для описания термодинамических свойств воды, что ею широко пользуются — и мы тоже будем к ней обращаться. Но надо иметь в виду, что она не точна.
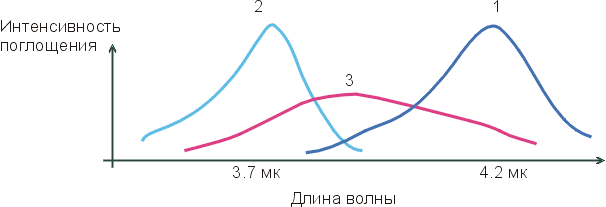
Рис.4-2. Характерный вид инфракрасных спектров поглощения О-Н групп во льду (1), в окружении CCl4 (2) и в жидкой воде (3).
Обратимся теперь к взаимодействию белковой цепи с водой.
Как и вода, остов белковой цепи полярен. Точнее — полярны входящие в остов пептидные группы. В целом остов белковой цепи имеет заряд 0, но распределение зарядов на его атомах (здесь опять заряд каждого атома выражен в долях от протонного заряда) выглядит так:
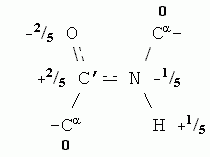
Парциальные заряды содержат и некоторые боковые группы, например, Ser (его боковая группа — - СН2-ОН — похожа на спирт). Еще более полярны заряженные аминокислотные остатки: заряд +1 присущ основаниям, входящим в боковые группы остатков Arg, Lys и His, когда они находятся в ионизованной форме (при примерно нейтральных рН), а у ионизованных кислотных боковых групп, Asp и Glu, заряд равен -1.
И пептидные группы главной цепи, и полярные боковые группы выступают донорами и акцепторами водородных связей. Они могут завязывать водородные связи друг с другом или с молекулами воды — и практически все они завязывают такие связи, так как энергия водородных связей — 5 ккал/моль — раз в десять превосходит энергию теплового движения, т. е. тепловое движение не может разрушить эти связи — разве что время от времени оно случайно нарушает малую часть из них.
Если внутримолекулярная связь между донором (D) и акцептором (А) водородной связи в белке завязывается в водном окружении, то эта связь замещает две водородные связи белка с молекулами воды (которые были до того, т. к. свято место пусто не бывает); и при этом еще создается одна связь между оторвавшимися молекулами воды:
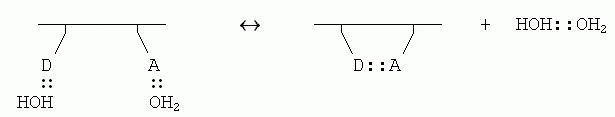
Энергетический баланс этой реакции близок к нулю: две Н-связи было, две стало. Однако энтропия воды возрастает, так как вода больше не привязана к белковой цепи и связанные Н-связью молекулы могут плавать где хочется (а энтропия именно и связана с множественностью состояний системы). При этом рост энтропии за счет освобождения димера воды от белка — примерно такой же, как рост энтропии при уходе молекулы Н2О изо льда в жидкую воду (в обоих случаях одна частица обретает свободу плавать).
Энтропийный эффект выхода молекулы Н2О изо льда в воду можно оценить следующим образом. Как известно, в точке плавления (для льда — при 0оС, т. е. при 273оК) энтропийный выигрыш от плавления как раз компенсирует энергетический проигрыш от него. А этот проигрыш нам известен: 80 кал/г, т. е. 80 Х 18 = 1440 кал/моль (18 - молекулярный вес Н2О).
Значит, вследствие роста энтропии свободная энергия системы "белок в воде" падает, — примерно на 1.5 килокалории на каждый моль формирующихся внутрибелковых водородных связей D:::A.
Эта выделенная водой свободная энергия может полностью или частично скомпенсировать рост свободной энергии, сопровождающий падение энтропии при фиксации групп D и А связью D:::A (раньше они болтались как хотели, а теперь, связанные, — не могут). Позже мы увидим, что падение свободной энергии воды примерно компенсирует энтропию необходимой для образования водородной связи (N-H:::O) фиксации мономера во вторичных структурах полипептидных цепей. В результате регулярная вторичная структура полипептидов в воде находится как раз на грани стабильности.
Итак, страшные слова — "энтропия" и "свободная энергия" — произнесены. Мой опыт показывает, что "энтропию" нормальный студент-биолог знает на слух (он слышал, что это — мера множественности состояний, мера беспорядка), а свободную энергию представляет совсем смутно... Так как нам понятие "свободная энергия" понадобится часто, я хочу уделить ей несколько минут сейчас, а потом, когда будет необходимо, поговорить о ней — и об энтропии — еще подробнее.
Начнем с простого примера: пусть молекула может находиться в двух состояниях, "а" и "б", например, — здесь, в этой комнате, на высоте 200 м от уровня моря ("а"), и в монастыре Далай-Ламы, в Гималаях, на высоте 5 км ("б"). Как соотносятся вероятности этих двух состояний — при условии, что мы следим за нашей молекулой так долго, что она успеет побывать и здесь, и там?
Обычно все помнят формулу Больцмана, которая гласит:
Вероятность быть в состоянии с энергией Е пропорциональна exp(-E/kT). | (4.2) |
Физический смысл этой формулы заключается в том, что теплота окружающей среды (столкновения с другими молекулами) в определенной степени (в среднем — пропорционально температуре среды T) возбуждает нашу молекулу, и это позволяет ей забираться в область более или менее высоких энергий. Все это мы подробнее обсудим позже, а пока я позволю себе надеяться, что вы эту формулу помните. Напомню только, что k — это постоянная Больцмана, а T — абсолютная температура в градусах Кельвина.
Применительно к интересующему нас вопросу — как соотносятся вероятности пребывания молекулы на разных высотах — формула Больцмана сводится к барометрическому соотношению
[Вероятность быть на высоте б] относится к [вероятности быть на высоте а] как exp(-Eб/kT) относится к exp(-Eа/kT) , | (4.3) |
где Eа — энергия молекулы в состоянии "а" (т. е. "здесь"), Eб — в состоянии "б" ("на высоте 5 км"), а T — абсолютная температура (для простоты, будем считать, что с высотой она не меняется).
Из-за силы тяжести энергия молекулы "здесь" ниже, чем "на высоте 5 км"; так что, согласно Больцману, "на высоте 5 км" молекула будет проводить меньше времени, чем "здесь" [раза в полтора-два меньше; посчитайте сами при T=300oK, вспомнив, что E=mgh, где m — средняя масса молекулы воздуха (»30 дальтон = 30 г/моль), g»10м/с2 — ускорение свободного падения, h»5км — разность высот, а постоянная Больцмана k равна 1.38x10-23 Джоуля/градус (Дж = кгxм2/сек2 = 0.24 кал) в расчете на частицу, т. е. 2 кал/град в расчете на моль (6x10-23) частиц. Напомню, что R = 2 (кал/моль)/град. называется "газовой постоянной"].
То есть молекул на высоте в 5 км будет раза в два поменьше, чем здесь. Точнее — поменьше в равных единицах объема, например — внутри легких (в чем легко убедиться, попробовав подышать на разных высотах). Однако в сумме число молекул в монастыре Далай-Ламы больше, чем в этой комнате: его монастырь куда больше, чем наша комната. То есть там молекула может находиться в большем числе положений — для свободно летающей молекулы воздуха это число пропорционально объему помещения. Физики говорят в этом случае, что в том монастыре возможно куда больше микросостояний молекулы, чем в нашей комнате. Поэтому вероятность того, что наша молекула находится где-то в монастыре Далай-Ламы, относится к вероятности того, что она находится где-то в этой комнате, как
[Вероятность где-то в объеме "б"] : [вероятность где-то в объеме "а"] =
= [Vбexp(-Eб/kT)] : [Vаexp(-Eа/kT)]
Здесь Vа — объем суммы состояний "а" ("нашей комнаты"), а Vб — объем суммы состояний "б" ("его монастыря"). Вспомнив элементарную институтскую математику, а именно, что V можно представить как exp(lnV), эту формулу можно переписать как
[Вероятность где-то в объеме б] : [вероятность где-то в объеме а] =
= [exp(-Eб/kT + lnVб)] : [exp(-Eа/kT + lnVа)] =
= [exp(-(Eб — Txk lnVб)/kT)] : [exp(-(Eа — Txk lnVа)/kT)]
Последняя формула очень напоминает формулу (4.3), формулу Больцмана, но только она относится не к единице объема, а ко всему объему системы, и — внимание! — вместо энергий Е в ней стоят величины E — Txk lnV. Так вот, величина F = E — Txk lnV называется свободной энергией нашей молекулы воздуха при заданном объеме V и температуре T. А величина S = k lnV называется энтропией нашей молекулы в объеме V.
Это — в данном конкретном случае, когда "число доступных состояний" — это просто доступный для молекулы объем V (Vа — "нашей комнаты", Vб — "его монастыря").
В общем случае, энтропия S просто равна k x [логарифм числа доступных состояний]. А свободная энергия F связана с энергией Е, энтропией S и температурой Т системы согласно общей формуле
F = E — TS | (4.4) |
Из двух состояний стабильнее, т. е. вероятнее то, у которого свободная энергия ниже:
[Вероятность_где-то_в_объеме_"б"] : [вероятность_где-то_в_объеме_"а"] = = exp(-Fб/kT) : exp(-Fа/kT) = exp[-(Fб-Fа)/kT] | (4.5) |
Иными словами, более стабильному состоянию системы отвечает то, где ниже F, а самому стабильному состоянию системы (при заданной температуре и фиксированном объеме этой системы) отвечает минимум свободной энергии F.
Таким образом — "свободная энергия" — это естественное расширение обычного понятия "энергии" на случай, когда система обменивается теплом с окружающей средой. Напомню: если тело не обменивается теплом со средой, его стабильное состояние отвечает минимуму его энергии (или, проще: все, что может упасть — падает). А окружающее тепло делает стабильными определенные флуктуации, прыжки — такие, которые минимизируют свободную энергию тела. То есть приток тепла возбуждает молекулы системы, они начинают захватывать многочисленные состояния с более высокой энергией (т. е. их энтропия растет) — и, в результате, молекулы воздуха летают, а не падают на землю.
Спустимся, однако, с Гималаев к белкам.
Итак — каков же баланс энергий, энтропий и свободных энергий в разобранном нами примере образования водородной связи в белковой цепи? Для полной наглядности, сравним ход процесса в разных условиях:
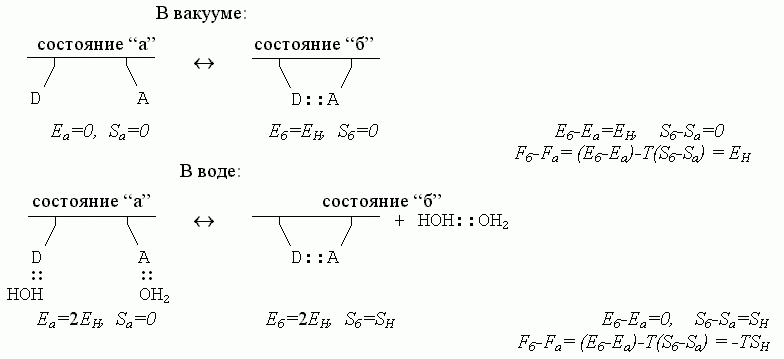
Здесь EH < 0 - энергия Н-связи, а SH > 0 — энтропия перемещений и вращения свободного тела, т. е. свободной молекулы Н2О или свободного димера НОH::OH2. Водородные связи — воды с водой, воды с белком — стабильны, когда EH -TSH < 0 (а если EH -TSH > 0, т. е. если они нестабильны, — то есть в этом случае перед нами была бы уже не жидкая вода, а пар).
Сравнение приведенных выше схем показывает, что водородные связи в белковой цепи как бы становятся — за счет изменения энтропии связывающейся воды — менее стабильными в водном окружении, чем в вакууме; действительно, в водном окружении их свободная энергия есть Fб-Fа = - ТSH , — то есть она меньше, чем в вакууме, где Fб-Fа=EH.
Еще раз хочу подчеркнуть, что причина этого видимого ослабления — в том, что в водном окружении образовавшаяся внутри белковой цепи Н-связь замещает собой связь цепи с водой. И по той же причине водородные связи, стабилизующие структуру белка в воде носят энтропийную, а не энергетическую природу: энергии двух состояний цепи (с внутрицепочечной связью и без нее) примерно равны, и из этих двух состояний с примерно равной энергией стабильнее та, где выше энтропия, т. е. где больше число микросостояний. А их больше у оторванной молекулы воды, чем у связанной.
Обратите внимание: водородные связи в белковой цепи (в водном окружении) носят энтропийную, а не энергетическую природу именно потому, что энергия Н-связей очень высока: раз так, "свободные" от связей в белке доноры и акцепторы водородных связей в цепи всегда на деле не свободны от всех связей вообще, а связаны с водой. Оторвавшиеся же от белка — при образовании внутрибелковой Н-связи — молекулы Н2О тут же связываются друг с другом, так что энергия компенсируется, и свободно-энергетический выигрыш Н-связей в белке идет только от множественности возможных микросостояний оторвавшихся молекул воды. Правда, чтобы связаться друг с другом, молекулы Н2О должны пожертвовать часть своей обретенной свободы, часть энтропии — но лучше уж потерять небольшую энтропию, чем большую энергию!
То, что (1) молекулы воды сильно связаны друг с другом водородными связями; и (2) то, что эти связи образуются только при определенной взаимной ориентации молекул Н2О — определяет специфику воды как растворителя. Это приводит к разным интересным эффектам. Подробнее мы об этом поговорим в двух следующих лекциях.
Лекция 5
В первой части этой лекции я хочу продолжить разговор о термодинамике. Это нам пригодится, когда мы будем потом обсуждать специфику воды как растворителя. Свободная энергия погружения разных молекул в воду играет в этом обсуждении ключевую роль.
Для изучения свободной энергии погружения молекулы в воду берут закупоренную пробирку, наполовину заполненную водой и наполовину — паром, воздухом или органическим растворителем, и смотрят, как изучаемые молекулы распределились между этими двумя фазами.
Мы уже видели, что разность свободных энергий F определяет предпочтительное состояние рассматриваемой системы (в данном случае — предпочтительное местонахождение рассматриваемой частицы) — согласно формуле
[Вероятность быть в б]:[вероятность быть в а] = exp[-(Fб-Fа)/kT] | (5.1) |
Свободная энергия F = E — TS складывается из энергии системы E и ее энтропии S. С энергией все, видимо, понятно. С температурой — понятно скорее интуитивно, и к ней мы еще вернемся. С энтропией — сложнее, и хочется повторить еще раз.
В простейшем случае частицы в сосуде, который мы, если вы помните, рассмотрели, S = k lnV, где V — доступный для частицы объем.
В чем смысл вхождения в свободную энергию этой энтропии (в виде члена -TS)? В том, что — если посмотреть на exp[-F/kT], забыв о E — то: exp[-(-TS)/kT] = exp[-(-TklnV)/kT] = V есть просто доступный объем, а он определяет число возможных состояний частицы в пространстве. Чем выше энтропия, тем больше это число состояний, тем вероятнее поэтому нахождение частицы именно в этом объеме.
В общем случае (когда возможные состояния частицы ограничены не только ее столкновениями со стенками, окружающими объем V, но и, скажем, столкновениями c другими частицами), энтропия S частицы равна
S = k´ [логарифм числа доступных для рассматриваемой частицы состояний].
В молекулярной физике, биологии, химии обычно говорят не об одной частице, а о моле (6 x 1023) частиц, и пишут
S = R´ [логарифм числа доступных для одной частицы состояний].
Здесь стоит еще раз напомнить, что различие между k и R — только в том, что k употребляют, говоря об одной частице, а R — о моле частиц.
Величина
F = E — TS | (5.2) |
называется, строго говоря, свободной энергией Гельмгольца. О ней просто рассказывать, и ее просто и удобно вычислять, так как эта величина относится к системе, которая (как та молекула, о которой мы говорили) находится в замкнутом и неизменном объеме.
Однако обычно на опыте измерения ведутся не при постоянном объеме V, а при постоянном внешнем давлении P (например — при атмосферном давлении). В этом случае измеряется не изменение чистой энергии Е изучаемого вещества, а изменение его энтальпии Н=Е+PV: в изменение энтальпии H, помимо собственно изменения энергии тела (Е), входит работа, совершенная против постоянного внешнего давления P при изменении объема V этого тела.
На практике — той, с которой мы будем иметь дело по ходу этих лекций — величину PV можно игнорировать, так как нас будут интересовать жидкости или твердые тела (где на каждую молекулу приходится небольшой объем), причем при невысоком (типа атмосферного) давлении. При этих условиях величина PV много-много меньше, чем тепловая энергия тела.
Поэтому для нас будет в дальнейшем несущественна разница между H и E. Обе эти величины я буду обычно называть просто энергия.
Столь же несущественно для нас будет различие между свободной энергией Гельмгольца F = E — TS и свободной энергией Гиббса
G = Н — TS = (E+PV) — TS = F + PV . | (5.3) |
И то, и другое (и G, и F) мы, не делая различия, будем именовать просто свободной энергией.
Пожалуй, стоит запомнить, что, обсуждая процессы, протекающие в замкнутом объеме, следует употреблять буквы E и F, а обсуждая процессы, протекающие при постоянном внешнем давлении — буквы H и G.
И еще следует запомнить то, что равновесному, стабильному состоянию любой системы при заданной температуре (T=const) соответствует минимум величины F=E - TS, если равновесие устанавливается при фиксированном объеме, и минимум величины G = H- TS, если оно устанавливается при фиксированном внешнем давлении.
При малом изменении состояния системы ее свободная энергия слегка меняется:
F ® F + dF = F + (dE - TdS - SdT) | |
или | (5.4) |
G ® G + dG = G + (dH - TdS - SdT) |
Это значит, в частности, что все возможные перестройки системы в окрестности ее стабильного состояния (а они могут изменять и энергию системы, и ее энтропию) подчиняются следующим уравнениям, характеризующим минимум функции (как мы знаем, точка минимума функции обладает тем свойством, что небольшое отклонение от нее практически не меняет функцию):
1) при V=const стабильному состоянию (при заданном Т) соответствует минимум F, т. е.
dF |V=const = dE |V=const - T dS |V=const = 0 | (5.5) |
[здесь мы учли, что dT=0 при T=const, т. е. SdT=0 в выражении (5.4)];
2) при P=const, когда стабильному состоянию (при заданном Т) соответствует минимум G,
dG |P=const = dH |P=const - T dS |P=const = 0. | (5.6) |
Из этих уравнений следует термодинамическое определение температуры:
T = (dE/dS) |V=const = (dH/dS) |P=const | (5.7) |
Я понимаю, что сейчас это определение получено довольно формально, и его физический смысл не очевиден. Поэтому я вернусь к этому вопросу в одной из ближайших лекций.
Остановимся теперь на химическом потенциале — величине, определяющей термодинамические характеристики не системы в целом, а одной молекулы в этой системе.
Если добавлять в систему молекулу за молекулой при постоянном давлении, то на добавление каждой новой частицы надо затратить в точности ту же работу, что на добавление любой предыдущей: объем системы будет расти, а плотность системы — и интенсивность взаимодействий в ней — меняться не будет. Поэтому термодинамическое состояние молекулы в системе удобно определять величиной свободной энергии Гиббса G, деленной на число молекул N,
m = G/N , | (5.8) |
называемой химическим потенциалом (а так как в жидкой или твердой фазе и невысоких давлениях F » G, то здесь m»F/N). Если N означает не число молекул, а, как обычно, число молей молекул, то и m отностися не к одной молекуле, а к молю молекул.
Химический потенциал — или, что то же самое, свободная энергия Гиббса в расчете на одну молекулу — нам пригодится во второй части сегодняшней лекции, когда речь пойдет о распределении молекул между фазами. Дело в том, что молекулы перетекают из той фазы, где их химический потенциал выше, в ту, где их химический потенциал ниже, — это понижает общую свободную энергию системы и приближает ее к равновесию. А в равновесии химический потенциал молекул в одной фазе равен химическому потенциалу тех же молекул в другой фазе.
Для будущего нам еще понадобится две формулы.
Во-первых, — определение теплоемкости, показывающей рост энергии системы с температурой:
CР= dН/dT |P=const | (5.9) |
(это — при постоянном давлении; можно считать теплоемкость и при постоянном объеме, но эти тонкости нам не нужны).
Во-вторых, — связь энтропии сo свободной энергией:
S = — dG/dT |P=const= — dF/dT |V=const | (5.10) |
Формулы (5.10) — важнейшие формулы термодинамики. Они прямо следует из того, что малое приращение свободной энергии dG = d(H-TS) = dH-TdS-SdT (и dF = d(E-TS) = dE-TdS-SdT), в то время как — в равновесии системы — dH-TdS=0 (и dЕ-TdS=0) в силу термодинамического определения температуры: T = dH/dS = dE/dS [см. (5.7)].
Формулы (5.10) показывают, что свободная энергия достигает своего минимального (или максимального) значения при той температуре T, когда S(T)=0.
Полезно знать также, что величина G/RT достигает своего минимального (или максимального) значения, когда H(T)=0, так как
d(G/RT)/dT = (dG/dT)/RT — G/RT2 = -S/RT — (H-TS)/RT2 = -H/RT2. | (5.11) |
После этого вступления мы готовы поговорить о специфике воды как растворителя. Прежде всего займемся так называемыми гидрофобным эффектом.
Гидро_фобия — в переводе — водо_боязнь. Кто же боится воды? Оказывается — все неполярные молекулы: и инертные газы (аргон, ксенон), и водород, и все чисто углеводородные молекулы (метан, этан, бензол, циклогексан и т. д.). Именно поведение углеводородов в воде нас и будет занимать: ведь в белках есть много аминокислот с углеводородными боковыми группами. И именно эти группы, боясь воды и убегая от нее, образуют гидрофобное ядро белковой глобулы.
Итак, в чем же — экспериментально - заключается гидро_фобия? Если мы измерим концентрацию метана — СН4 — в воде и в паре над ней, то в паре концентрация метана будет раз в 10 больше (точное число зависит от температуры, но при комнатной температуре, около 20-40оС, — примерно в 10 раз). Приблизительно то же соотношение будет и для Н2, и для пропана — СН3СН2СН3: все они боятся (фобия!) смешиваться с водой, и всех их больше в паре, чем в воде. А вот этиловый спирт СН3СН2ОН и вода хорошо смешиваются, и разделить их довольно трудно, — это все знают; но спирт — полярная молекула (ее полярная О-Н нруппа способна завязывать Н-связи). А чисто неполярным молекулам лучше уж в вакууме (пар — это почти вакуум), чем в воде... И это — несмотря на Вандерваальсово притяжение любых молекул — в том числе Н2 или СН4 к Н2О!
Рассмотрим на одном примере (Рис.5-1) характерные термодинамические эффекты растворения углеводородов в воде. Приведенные на Рис.5-1 термодинамические данные (свободные энергии, энергии и энтропии переноса из фазы в фазу) основаны на изучении равновесного распределения молекул циклогексана, (СН2)6, между тремя фазами: паром, водным раствором и жидким циклогексаном.
Левая часть Рис.5-1 показывает, что, при 25оС, пар циклогексана, имеющий стандартное давление в 1 атм., уменьшает свою свободную энергию на DG = DGгаз®жидкий_циклогексан = -2 ккал/моль при конденсации в жидкий циклогексан. Это значит, что при давлении в 1 атм. пар циклогексана перенасыщен (раз он уменьшает свою свободную энергию при конденсации), а насыщенный (т. е. находящийся в равновесии с жидкой фазой) пар циклогексана, (СН2)6, при 25оС имеет давление, отличеющееся в ехр[-(DGжидкий_циклогексан®газ)/RT] раз от его стандартного давления (1атм.), относительно которого отсчитываются все термодинамические функции газа. То есть насыщенный пар циклогексана имеет давление в
1атм x ехр[-(DGжидкий_циклогексан®газ)/RT] = 1атм x ехр[+(DG газ®жидкий_циклогексан)/RT] »
» 1атм x ехр[(-2)/0.6] » (1/28) атм.
А так как плотность газа пропорциональна его давлению, и так как моль газа при атмосферном давлении (и комнатной температуре) занимает около 25 литров, то моль насыщенного пара (СН2)6 занимает литров 700. То есть концентрация (СН2)6 в насыщенном паре — около (1/700) моль/литр.
Нижняя часть Рис.5-1 показывает, что насыщенный раствор (СН2)6в воде имеет концентрацию 1моль/литр x ехр[-(DGжидкий_циклогексан®вода)/RT] » ехр[-(+6)/0.6] моль/литр » 1/20000 моль/литр. [Напомню, что состояние раствора отсчитывается от его стандартной концентрации в 1 моль/литр]. То есть концентрация (СН2)6 в воде, (1/20000) моль/литр, — раз в 30 меньше его концентрации в паре.
О том же соотношении концентраций 30:1 в пользу пара говорит и правая, замыкающая термодинамический цикл часть Рис.5-1. Она говорит, что концентрации растворенного в воде и газообразного (СН2)6 соотносятся как

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 |
