
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Я не буду утомлять вас подробным решением всей задачи, а сразу напишу ответ. Итак.
Поле над белком создается и зарядом q, и отраженным зарядом q'= q (e1- e2)/ (e1+ e2) » +q, лежащим на той же глубине под поверхностью, как q — над поверхностью белка (Рис.6-5).
| Рис.6-5. |
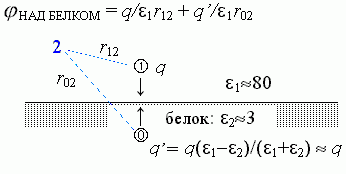
В результате, эффективная диэлектрическая проницаемость eэфф (это — та проницаемость, которую надо подставлять в формулу j = q/eэффr12), — эта eэфф в зоне над белком близка к e1, т. е. к 80 только на самых малых расстояниях от заряда ![]() (когда r12 много меньше, чем расстояние от
(когда r12 много меньше, чем расстояние от ![]() до поверхности белка; при этом r12 << r02»r01).
до поверхности белка; при этом r12 << r02»r01).
Но на больших расстояниях от ![]() , — здесь eэфф всюду близка к 40. В самом деле, когда r12>>r10, то r02 » r12, и так как q'» q при e1>>e2 , то j » 2q/[er12]= q/[(e1/2)r12].
, — здесь eэфф всюду близка к 40. В самом деле, когда r12>>r10, то r02 » r12, и так как q'» q при e1>>e2 , то j » 2q/[er12]= q/[(e1/2)r12].
При этом eэфф » 40 относится и к полю над (а следовательно — и под) сaмой поверхностью белка (где всегда r02 » r12, см. Рис.6-5).
Итак, поле внутри белка создается зарядом, стоящим в точке "1" (Рис.6-6), и для этого поля — я опять позволю себе опустить выкладки — эффективная диэлектрическая проницаемость во всем пространстве под поверхностью равна eэфф =(e1+e2)/2, т. е. близка к 40.
| Рис.6-6. |
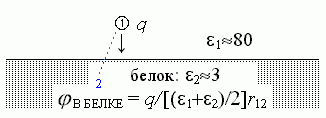
Очень похожий ответ получится, если мы будем рассматривать комплементарную задачу: о поле заряда, находящегося под поверхностью белка (Рис.6-7).
| Рис.6-7. |

В этом случае эффективная диэлектрическая проницаемость eэфф тоже близка к (e1+e2)/2, т. е. к 40 всюду на больших расстояниях от заряда q, а к e2 (т. е. к 3), — только когда r12 много меньше, чем расстояние от заряда q до поверхности белка.
Еще более любопытный результат дает задача о поле, которое заряд, находящийся на одном краю белка, создает на другом краю белка (Рис.6-8).
| Рис.6-8. |
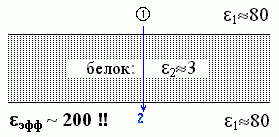
Казалось бы, в этом случае, поскольку взаимодействие идет через белок, через среду с низкой (e2»3) диэлектрической проницаемостью, — мы должны были бы ожидать, что eэфф будет близко к 3, в крайнем случае — ведь вода вокруг — лежать где-то между 3 и 80. А на самом деле — eэфф зашкаливает за 200! Как так??
Для объяснения посмотрим, как ориентируются в этом случае полярные молекулы воды вокруг белка и вокруг заряда ![]() , и как это меняет поле заряда (см. Рис.6-9; он дан, для наглядности, в несколько другом масштабе, чем Рис.6-8, но описывает то же явление).
, и как это меняет поле заряда (см. Рис.6-9; он дан, для наглядности, в несколько другом масштабе, чем Рис.6-8, но описывает то же явление).
Рис.6-9. Ориентация молекул воды (они изображены в виде диполей |
|
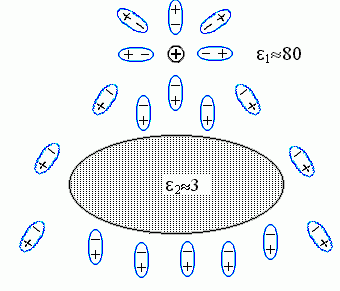
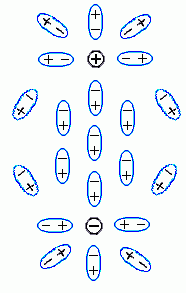
Молекулы воды ориентируются по полю: их "-" преимущественно смотрит в сторону заряда ![]() , а "+" — в противоположную сторону. В результате, во-первых, заряд частично компенсируется примыкающими к нему "минусами" молекул воды; это — тривиальное явление, оно просто приводит к появлению диэлектрической проницаемости воды, e1. Во-вторых — возникает то явление, которое нас интересует. А именно, — повернутые к белку "плюсы" молекул воды, примыкающих к белку с той стороны, где находится заряд
, а "+" — в противоположную сторону. В результате, во-первых, заряд частично компенсируется примыкающими к нему "минусами" молекул воды; это — тривиальное явление, оно просто приводит к появлению диэлектрической проницаемости воды, e1. Во-вторых — возникает то явление, которое нас интересует. А именно, — повернутые к белку "плюсы" молекул воды, примыкающих к белку с той стороны, где находится заряд ![]() , создают на этой стороне поляризационный заряд "+"; а "минусы" молекул воды, примыкающих к белку с другой стороны, создают там противоположный поляризационный заряд "-" (поляризацией самого тела белка можно пренебречь, — по сравнению с поляризацией воды она мала, так как его e2 <<e1).
, создают на этой стороне поляризационный заряд "+"; а "минусы" молекул воды, примыкающих к белку с другой стороны, создают там противоположный поляризационный заряд "-" (поляризацией самого тела белка можно пренебречь, — по сравнению с поляризацией воды она мала, так как его e2 <<e1).
Сумма поляризационных зарядов у поверхности исходно незаряженного тела, окруженного однородной средой (у нас — водой), должна быть равна нулю — есть такая теорема в электростатике. Значит, суммарный поляризационный заряд "+" у поверхности белка обращен к ![]() (!!), а равный ему поляризационный "-" (у другой его поверхности), — в обратную сторону.
(!!), а равный ему поляризационный "-" (у другой его поверхности), — в обратную сторону.
В результате на обращенной к заряду стороне белка потенциал поля возрастает по сравнению с тем, что было бы без белка, — здесь, в дополнение к потенциалу заряда ![]() , потенциал создается и поляризационными "плюсами" у этой поверхности белка (а поляризационные "минусы" от этого места далеко и влияют мало). Поэтому здесь eэфф»40, как о том уже говорилось.
, потенциал создается и поляризационными "плюсами" у этой поверхности белка (а поляризационные "минусы" от этого места далеко и влияют мало). Поэтому здесь eэфф»40, как о том уже говорилось.
В то же время, на отвернутой от заряда стороне белка потенциал поля падает по сравнению с тем, что было бы без белка, — здесь к потенциалу заряда ![]() добавляется противоположный по знаку потенциал поляризационных "минусов" у ближайшей поверхности белка (а поляризационные "плюсы" от этого места далеко и влияют мало). А раз здесь потенциал поля упал по сравнению с тем, что было бы без белка, — то здесь (на противоположной
добавляется противоположный по знаку потенциал поляризационных "минусов" у ближайшей поверхности белка (а поляризационные "плюсы" от этого места далеко и влияют мало). А раз здесь потенциал поля упал по сравнению с тем, что было бы без белка, — то здесь (на противоположной ![]() 'у стороне) eэффбольше, чем то e1, что было без белка, — т. е. больше, чем e1=80.
'у стороне) eэффбольше, чем то e1, что было без белка, — т. е. больше, чем e1=80.
В целом, распределение значений eэфф "в белке и вокруг" для поля, создаваемого зарядом ![]() и наведенного им же поляризационными зарядами, выглядит так, как на Рис.6-10, если заряд
и наведенного им же поляризационными зарядами, выглядит так, как на Рис.6-10, если заряд ![]() находится у поверхности белка, и так, как на Рис.6-11, если заряд
находится у поверхности белка, и так, как на Рис.6-11, если заряд ![]() находится в глубине белка.
находится в глубине белка.
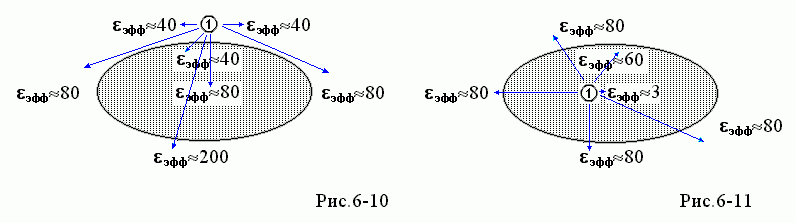
Напоминаю, что eэфф — это та эффективная величина диэлектрической проницаемости для точки r, которую нужно подставлять в формулу расчета потенциала заряда "1" в точке r: f(r)= q1/eэфф|r-r1|.
Отметим еще одно явление, вытекающее из наличия поверхности раздела и связанных с ней "отраженных зарядов". Оно касается действия заряда на самого себя [см. также обсуждение формулы (6.3)]: находящийся вне белка заряд отталкивается от поверхности белка, а заряд, находящийся внутри белка, сильно притягивается к его поверхности — т. е., в обоих случаях, среда с более высокой диэлектрической проницаемостью притягивает заряд, а среда с более низкой — его выталкивает. Силу этого притяжения и выталкивания легко оценить — ведь речь здесь идет о взаимодействии заряда с его отражениями (см. Рис.6-5, 6-6), но в этом я рекомендую вам попрактиковаться самостоятельно.
Теперь займемся эффектами, связанными с корпускулярностью, т. е. атомарным строением среды.
Собственно говоря, сама величина диэлектрической проницаемости e определяется атомарным строением среды. Если среда состоит из неполярных молекул, электрическое поле лишь смещает электроны в них, что сделать трудно: поэтому электроны смещаются мало, и e невелико. Если среда состоит из полярных молекул (пример — вода), электрическое поле разворачивает эти молекулы; это сделать легче, и e такой среды велико. В обоих случаях — и при смещении электронов, и при повороте полярных молекул, — поляризация среды как бы частично "гасит" внесенные в нее заряды (заряды ![]() и
и ![]() , Рис.6-12), и тем самым уменьшает электрическое поле в среде, — по сравнению с тем, что было бы в вакууме.
, Рис.6-12), и тем самым уменьшает электрическое поле в среде, — по сравнению с тем, что было бы в вакууме.
| Рис.6-12. |
Естественно было бы предположить, что связанные с корпускулярностью эффекты должны сильно влиять на взаимодействие зарядов на малых расстояниях — ведь классические формулы (6.1 — 6.3) справедливы, строго говоря, только когда между взаимодействующими зарядами находится много молекул среды. А если заряды (как часто бывает в белках) находятся на расстоянии 3-4 ![]() , никакой другой атом между ними уже не влезет и не изменит их взаимодействия.
, никакой другой атом между ними уже не влезет и не изменит их взаимодействия.
Казалось бы, в случае столь тесного контакта зарядов — диэлектрическая проницаемость для их взаимодействия должна приближаться к 1 даже в водном окружении. Эту точку зрения — точнее, это опасение — до сих пор можно встретить в литературе.
Однако, как ни странно, корпускулярность среды не меняет кардинально "макроскопическую" (т. е. выведенную для больших расстояний между зарядами) диэлектрическую проницаемость среды даже на расстоянии порядка 3 ![]() . То есть даже здесь eэфф гораздо ближе к 80 или 40, чем к 1 или 3.
. То есть даже здесь eэфф гораздо ближе к 80 или 40, чем к 1 или 3.
Об этом свидетельствует то, что соль хорошо растворяется (диссоциирует) в воде, что возможно только при слабом притяжении противоионов даже на самых малых, »3![]() расстояниях.
расстояниях.
В самом деле, ионы Na+ и Cl - могут сблизиться до расстояния 3![]() . При этом свободная энергия их притяжения составляла бы —1.5 ккал/моль при e=80, —3 ккал/моль при e=40, и —6 ккал/моль при e=20. Последнее (6 ккал/моль) превосходит энергию водородной связи. При такой энергии противоионы слипались бы друг с другом сильнее, чем молекулы воды, и тогда насыщенный раствор соли имел бы концентрацию порядка 10-4 моля на литр — как насыщенный водный пар. Но это явно неправда: растворить 1 моль NaCl (58 г) в литре воды — не проблема (это — обычный, может чуть крепкий рассол). Значит, диэлектрическая проницаемость воды существенно больше 20 даже на расстоянии »3
. При этом свободная энергия их притяжения составляла бы —1.5 ккал/моль при e=80, —3 ккал/моль при e=40, и —6 ккал/моль при e=20. Последнее (6 ккал/моль) превосходит энергию водородной связи. При такой энергии противоионы слипались бы друг с другом сильнее, чем молекулы воды, и тогда насыщенный раствор соли имел бы концентрацию порядка 10-4 моля на литр — как насыщенный водный пар. Но это явно неправда: растворить 1 моль NaCl (58 г) в литре воды — не проблема (это — обычный, может чуть крепкий рассол). Значит, диэлектрическая проницаемость воды существенно больше 20 даже на расстоянии »3![]() .
.
Более точно оценить величину e на самых малых близких расстояниях внутри молекулы можно, исходя из величин первой и второй констант диссоциации двухосновных кислот и оснований в воде. Например, диссоциация щавелевой кислоты происходит так:
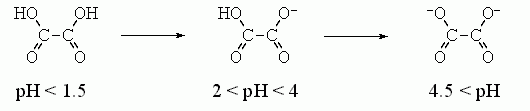
Вторая диссоциация сдвинута на »2.5 единицы рН относительно первой, т. е. происходит при в 102.5 = е2.3x2.5 раз меньшей концентрации Н+. Это показывает, что свободная энергия взаимодействия первого заряда со вторым составляет 2.5x2.3RT » 3.5 ккал/моль. И это — при расстоянии в »3 ![]() между зарядами. Такая величина энергии взаимодействия соответствует e»30?40 на расстоянии в 3
между зарядами. Такая величина энергии взаимодействия соответствует e»30?40 на расстоянии в 3 ![]() . К сходному результату — e» 30?40 на расстоянии в » 2-2.5
. К сходному результату — e» 30?40 на расстоянии в » 2-2.5 ![]() — приводит рассмотрение диссоциации угольной кислоты, Н2СО3® НСО3-® СО3--.
— приводит рассмотрение диссоциации угольной кислоты, Н2СО3® НСО3-® СО3--.
Значит, даже солевая связь противоположно заряженных боковых групп на поверхности белка должна "стоить" всего пару-тройку ккал/моль. Внутри белка она должна "стоить" больше, но погружение заряженных групп внутрь глобулы обойдется еще дороже — так что не надо удивляться, что такие связи в природных белках наблюдаются нечасто.
Итак, мы пришли к выводу, что корпускулярность не меняет радикально "макроскопическую" (выведенную для больших расстояний между зарядами) диэлектрическую проницаемость воды даже на расстоянии в 2 — 3 ![]() , когда между взаимодействующими зарядами не может быть никаких других молекул. Причина этого — в том, что заряды достаточно сильно экранируются даже подошедшими "с другой стороны и с боков" (Рис.6-13) молекулами среды, которые поляризуются (в случае воды — просто поворачиваются) так, что "+"'ы этих молекул смещается к заряду
, когда между взаимодействующими зарядами не может быть никаких других молекул. Причина этого — в том, что заряды достаточно сильно экранируются даже подошедшими "с другой стороны и с боков" (Рис.6-13) молекулами среды, которые поляризуются (в случае воды — просто поворачиваются) так, что "+"'ы этих молекул смещается к заряду ![]() , а "-"'ы — к заряду
, а "-"'ы — к заряду ![]() .
.
| Рис.6-13. |
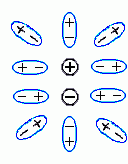
Здесь мы снова (ср. Рис.6-10, 6-11) видим, что электростатическое взаимодействие между зарядами (![]() и
и ![]() ) как бы проходит в основном через среду с более высокой диэлектрической проницаемостью и почти игнорирует среду, которая слабо поляризуется.
) как бы проходит в основном через среду с более высокой диэлектрической проницаемостью и почти игнорирует среду, которая слабо поляризуется.
Все, о чем мы говорили до сих пор, относилось к "мелкомолекулярным" системам. Сохраняются ли все эти выводы для белков?
Опыты, проведенные в лаборатории Фершта — зачинателя белковой инженерии — показывают, что приведенные выше оценки в полной мере относятся и к белкам.
Опыты основывались на следующем. Есть белки, ферменты, активность которых особенно велика при определенном значении рН (у них, как говорится, есть рН-оптимум).
Вводя в белок, путем мутации его гена, заряженный остаток, можно сдвинуть этот рН-оптимум (Рис.6-14) — и, по его сдвигу, оценить электрическое поле, созданное мутировавшим остатком в активном центре.
| Рис.6-14. |
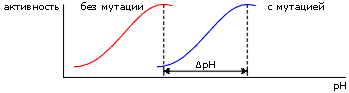
Причина рН-оптимума — в том, что какая-то группа в активном центре должна, для успешной работы фермента, находиться в определенной зарядовой форме, а заряженность этой группы зависит от концентрации водородных ионов в среде.
Пусть активный центр (АЦ) принимает ион Н+: АЦ + Н+ = АЦН+. Тогда, по закону действующих масс, соотношение концентраций двух (с Н+ и без Н+) форм активного центра составляет [АЦН+]/[АЦ] = exp(-DFАЦН+/RT) x [H+] = exp(-DFАЦН+/RT) x 10-рН = exp{-(DFАЦН+/RT + 2.3 x рН)}, где DFАЦН+ — свободная энергия связывания Н+ с активным центром, а значок [ ] означает концентрацию.
Если мутация вносит в белок заряд, создающий в его активном центре потенциал j, то DF АЦН+ меняется: DF АЦН+|с_мутацией = DFАЦН+ |без_мутации + je, где e — заряд Н+. Так как в рН-оптимуме величина [АЦН+]/[АЦ] (и, следовательно, величина DFАЦН+/RT + 2.3 x рН) должна быть одной и той же и при, и без мутации, — то DFАЦН+|без_мутации /RT + 2.3 x рН|опт._без_мутации = DFАЦН+|с_мутацией /RT + 2.3 x рН|опт._с_мутацией. То есть je = DFАЦН+|с_мутацией — DF АЦН+|без_мутации = 2.3RT(рН|опт._без_мутации — рН|опт._с_мутацией) = 2.3RTx(-DрН).
Так, зная сдвиг pH-оптимума, можно оценить потенциал, созданный в активном центре мутированным остатком белка. Теперь, зная трехмерную структуру этого белка и, следовательно, расстояние r от мутированного остатка до активного центра, — можно оценить эффективную диэлектрическую проницаемость eэфф (входящую в формулу j = q/eэффr) для взаимодействия внесенного мутацией заряда q с районом активного центра.
Мутации в опытах Фершта вводились на поверхность белка, чтобы не разрушить его структуру (мы уже знаем, что энергия глубоко погруженного в белок заряда очень велика, так что он может буквально взорвать белок).
Результат: эффективная диэлектрическая проницаемость eэфф колебалась в этих опытах от ~40 до ~120, причем первое характерно для близких к активному центру мутаций, а второе — для удаленных от него. Последнее — то, что eэфф может достигать 120 — вызвало изрядное удивление, так как многие, не знакомые как следует с электростатикой, считали, что eэфф должны лежать где-то между 3 (как внутри белка) и 80 (как в воде). Однако нас эти величины удивлять не должны — они хорошо согласуются с тем, что можно ожидать из Рис.6-10.
Небольшое отступление — о белковой инженерии. Ее главная прелесть заключается в том, что, меняя такой-то кодон в гене белка, мы можем ввести мутацию в точно определенное место белковой глобулы, так как и ген этого белка, и его аминокислотная последовательность, и его трехмерная структура известны. Кроме того, влияние мутации на структуру также можно контролировать — рентгеноструктурным анализом или ЯМР. Таким образом, вся работа ведется с открытыми глазами.
В опытах, о которых шла речь, белок служит микроскопическим (вернее, наноскопическим) электрометром. И белковая инженерия дает возможность использовать такие приборы — и при этом прыгать от физической теории к генным манипуляциям и обратно, что чрезвычайно занимательно.
Теперь я хочу сделать несколько добавлений касательно электростатических взаимодействий.
Первое. До сих пор я говорил только о взаимодействии отдельных зарядов. Однако к электростатике относятся и взаимодействия диполей (например, диполей Н(+)-О(-) и Н(+)-N(-), вовлеченных в образование водородных связей), а также квадруполей — последние присутствуют, например, в ароматических кольцах (Рис.6-15).
Рис.6-15. Электрический квадруполь ароматического кольца: |
|
Я остановился на взаимодействии зарядов потому только, что они наиболее сильны, — даже при прямом контакте они в несколько раз сильнее, чем взаимодействия диполей (и к тому же медленнее спадают с расстоянием), а взаимодействия диполей, — сильнее, чем взаимодействия квадруполей.
Второе. При наличии свободных зарядов (например, соли) в воде, электростатические взаимодействия ослабевают с расстоянием r не по закону "энергия пропорциональна (1/r)", а гораздо быстрее — как (1/r) x exp(-r/D). Здесь D — радиус Дебая-Хюккеля — соответствует характерному размеру противоионного облака вокруг заряда. Величина D не зависит от самого заряда, но зависит от концентрации ионов в среде, от ее диэлектрической проницаемости и температуры. В воде, при комнатной температуре,
D » 3/I1/2 | (6.5) |
где
I = 1/2 åi ci zi2 — | (6.6) |
ионная сила раствора в моль/литр. В формуле (6.6) сумма берется по всем сортам ионов в растворе, причем zi — заряд (в единицах протонного заряда), а ci — концентрация (в моль/литр) иона i. Обычным физиологическим условиям соответствует I » 0.1-0.15; при этом D » 8![]() . Однако некоторые микроорганизмы живут при I ~ 1 моль/л и выше; при этом сохраняются, и то в очень ослабленном виде, только те электростатические взаимодействия, что соответствуют "солевым мостикам", т. е. прямому контакту зарядов.
. Однако некоторые микроорганизмы живут при I ~ 1 моль/л и выше; при этом сохраняются, и то в очень ослабленном виде, только те электростатические взаимодействия, что соответствуют "солевым мостикам", т. е. прямому контакту зарядов.
В целом, при наличии в растворе ионной атмосферы, энергия взаимодействия двух зарядов имеет вид
U = [q1q2/eэффr] x exp(-r/D) . | (6.7) |
Третье. Электростатическое взаимодействие — яркий пример не парного взаимодействия частиц (в отличие от, например, Вандерваальсового). Оно зависит не только от расстояния r между зарядами q1 и q2, но и от свойств среды (меняющих и e, и D), и в частности — от расстояний от зарядов до других тел и от формы этих тел (все это влияет на eэфф).
И еще одно добавление. До сих пор я обычно говорил "энергия электростатических взаимодействий". Это говорилось только для простоты слога — как я уже говорил в начале лекции, строго говоря, надо было говорить "свободная энергия".
Более того. Исследуя температурную зависимость электростатических эффектов в водном окружении, можно показать, что энтропийная составляющая в них доминирует, а собственно энергетическая (энтальпийная) составляющая близка к нулю. Это видно из того, что диэлектрическая проницаемость воды меняется от 88 до 55 (т. е. электростатические взаимодействия растут примерно на 40%) при росте абсолютной температуры Т от 273 до 373оК (тоже примерно на 35%). А у взаимодействия, растущего пропорционально абсолютной температуре, есть только энтропийная, но нет энтальпийной части. Значит, в воде весь электростатический эффект связан не с энергией, а с упорядочением воды вокруг зарядов и с изменением этого упорядочения при сближении или отдалении зарядов друг от друга.
Значит, как ни парадоксально, а электростатика в воде имеет энтропийную, а не энергетическую природу, — впрочем, так же, как и гидрофобные взаимодействия или как и образование водородных связей в водном окружении.
Заключая раздел "Элементарные взаимодействия в белках и вокруг", я хочу еще упомянуть о дисульфидных связях и о координационных связях. Не столь массовые, как, например, водородные связи, — эти связи, однако, часто играют важную роль в белках.
Дисульфидные (или SS) связи образуются цистеиновыми (Cys) аминокислотными остатками (боковая группа цистеина: - CbH2-SH). Непосредственное, с выделением водорода (по схеме - CH2-SH + HS-CH2- ® - CH2-S-S-CH2- + H2) окисление цистеинов в белках не идет — оно происходит слишком медленно при комнатной температуре. Однако образование SS связей в белках может быстро происходить при помощи тиол-дисульфидного обмена. Полагают, что в клетке это происходит при участии глутатиона, существующего и в мономерной тиольной (GSH), и в димерной дисульфидной (GSSG) формах, — и происходит по схеме
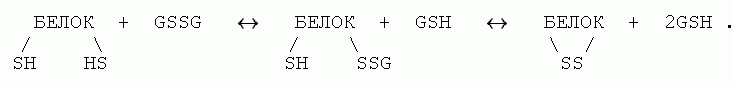
Как разрыв, так и образование SS связей в клетках катализируется (т. е. ускоряется, но не направляется) специальным ферментом — дисульфидизомеразой.
Образование S-S связей в клетке обратимо, так как энергетический баланс этой реакции — тиол - дисульфидного обмена — близок к нулю (было две ковалентные SH связи и одна SS связь, и столько же осталось; не правда ли, это очень походе на энергетический баланс образования внутрибелковых водородных связей в водном окружении?). Более того, имеющаяся — относительно высокая — концентрация GSH в клетке сдвигает равновесие в сторону разрыва тех связей, которые могли бы образоваться при "случайном" сближении цистеинов. Поэтому здесь способны образоваться и выжить SS связи только между "и без того" (т. е. — другими взаимодействиями) сближенными цистеинами.
Особенно важны SS связи для белков, которым придется жить и работать вне клетки. С одной стороны, там нет дисульфидизомераз и глутатиона, так что уже завязавшиеся (внутри клетки или на выходе из нее) связи "замораживаются" — они уже не порвутся и на перестроятся. С другой — белок вне клетки попадает в разные условия, и дополнительный запас прочности, даваемый стабильными замороженными SS связями, ему не помешает. Поэтому SS связи гораздо более типичны для секретированных белков, чем для внутриклеточных. Обычно в секретированных белках все имеющиеся цистеины (кроме одного, если их число нечетно) вовлечены в SS связи.
Координационные связи образуются N, O и S атомами белка (а также О атомами воды) с двух— и трехвалентными ионами металлов: Fe, Zn, Со, Ca, Mg и т. д.
У ионов этих металлов есть незаполненные, но низко лежащие орбитали. Каждая из них способна связать электронную пару. А у О, N, S атомов ("доноров электронов") есть электронные пары, которые могут перетечь на свободные орбитали ионов. Образующаяся при этом связь ничем не отличается от обычной химической связи — кроме того, что для обычной связи электроны поставляются обоими входящими в связь атомами, а для координационной — только одним из них.
При образовании координационных связей ион металла связывается с несколькими донорами электронов. При этом крупные (с радиусом ~1.5![]() ) атомы-доноры со всех сторон окружают маленький (с радиусом ~0.7
) атомы-доноры со всех сторон окружают маленький (с радиусом ~0.7![]() ) двух— или трехвалентный ион. Наиболее часто шесть атомов-доноров окружают (координируют) ион металла, располагаясь по вершинам правильного октаэдра (Рис.6-16).
) двух— или трехвалентный ион. Наиболее часто шесть атомов-доноров окружают (координируют) ион металла, располагаясь по вершинам правильного октаэдра (Рис.6-16).
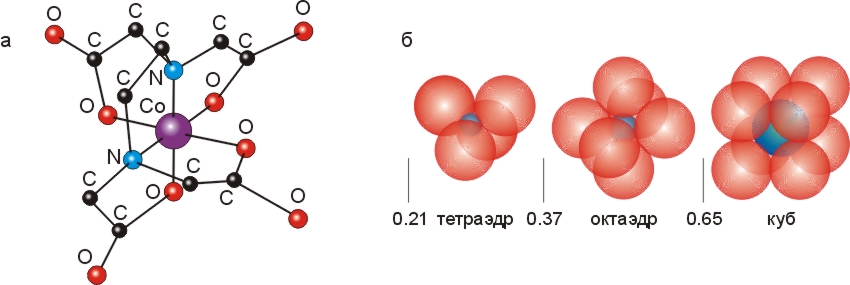
Рис.6-16. (а) Структура октаэдрического комплекса, образованного атомом Со+++ с EDTA. (б) Характерная координация центрального иона при различном соотношении его радиуса с радиусами окружающих его доноров электронов. Картинки взяты из [10] и адаптированы.
Так как ион может связаться и с электронными донорами белка, и с кислородами воды, он (несмотря на большую энергию каждой связи) переходит из воды на белок и обратно без драматического выигрыша или проигрыша энергии. Важнее, пожалуй, другое: если в белке атомы - доноры уже стоят в "правильных" (для образования координационной связи) положениях, — ион может связаться с ними, отпустив ранее окружавшие его молекулы воды, а это приведет к прочной связи из-за выигрыша энтропии движения отпущенных молекул воды. В среднем, каждая координационная связь стоит несколько ккал/моль — несколько больше, чем водородная связь в воде.
Такие связи, образуемые несколькими атомами одной молекулы, способными связать один ион, называются хелатными ("клешневидными").
Роль таких связей в белках, и в частности в их активных центрах, мы рассмотрим позже. Мы увидим также, что хелатные, полностью обволакивающие ион комплексы могут входить в гидрофобное ядро белка. А пока я хочу снова обратить ваше внимание на Рис.6-16, где изображен часто используемый в лабораторной практике реагент ЭДТА (этилендиаминтетраацетат) в хелатной связи с металлом. У ЭДТА эта связь особенно прочна из-за связи отрицательных зарядов СОО- групп ЭДТА с положительно заряженным ионом металла.
Лекция 7
Разобравшись с элементарными взаимодействиями, — рассмотрим сегодня вторичную структуру белков. Прежде всего у нас речь пойдет о регулярных вторичных структурах — об a-спиралях и о b-структуре, — но не только о них.
Укладка a и b-структур в глобулу определяет третичную структуру белка (Рис.7-1). Эти вторичные структуры отличаются определенными, периодическими конформациями главной цепи — при разнообразии конформаций боковых групп.
Рис.7-1. Вторичная структура полипептидной цепи (a-спираль и тяж b-листа) и третичная структура белковой глобулы.
Начнем со спиралей. Они могут быть левые и правые (Рис.7-2), у них может быть разный период и шаг. Правые (R) спирали приходят к нам, завиваясь против часовой стрелки (что отвечает положительному отсчету угла в тригонометрии); левые (L) — приходят, вращаясь по стрелке.
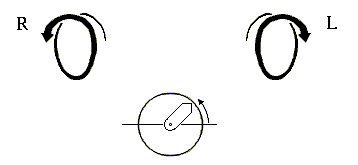
Рис.7-2. Правые (R) и левые (L) спирали. Под ними показан отсчет положительного угла в тригонометрии: при этом "близкая к нам" стрелка вращается против хода часов.
Важнейшие спирали в полипептидной цепи держатся водородными связями, где С=О группы остова полипептида связаны с лежащими от них в направлении С-конца цепи H-N группами. В принципе, возможны следующие спирали, стянутые Н-связями (Рис.7-3): 27, 310, 413 (обычно именуемая a) и 516 (она же p). Здесь в названии "27" — "2" означает связь со 2-м по цепи остатком (см. Рис.7-3), а "7" — число атомов в цикле (O......H-N-C'-Ca-N-C'), замыкаемом этой связью. Тот же смысл имеют цифры и в названии других спиралей.
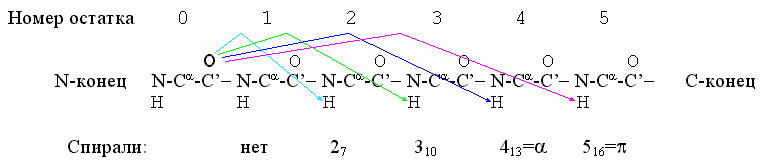
Рис.7-3. Водородные связи (они показаны стрелками), характерные для разных спиралей.
Какие из этих спиральных структур преобладают в белках? a-спирали. Почему? Ответ на этот вопрос дает карта Рамачандрана для типичного аминокислотного остатка — аланина (Рис.7- 4), на которой отмечены конформации, периодическое повторение которых приводит к завязыванию изображенных на Рис.7-3 водородных связей.
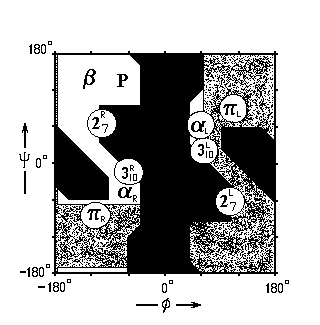
Рис.7-4. Конформации различных вторичных структур на фоне карты разрешенных и запрещенных конформаций аминокислотных остатков. 27R, 27L: правая и левая спираль 27; 310R, 310L: правая и левая спираль 310; aR, aL — правая и левая a-спираль; pR, pL — правая и левая p-спираль. b — b-структура (подробности см. на Рис.7-8б). Р — спираль Poly(Pro)II. ![]() — конформации, разрешенные для аланина (Ala);
— конформации, разрешенные для аланина (Ala); ![]() — области, разрешенные лишь для глицина, но не для аланина и других остатков;
— области, разрешенные лишь для глицина, но не для аланина и других остатков; ![]() — области, запрещенные для всех остатков. j и y — углы внутреннего вращения в белковой цепи.
— области, запрещенные для всех остатков. j и y — углы внутреннего вращения в белковой цепи.

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 |
