
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Статистическая сумма играет очень важную роль в статистической физике, так как величина Z(Т) позволяет прямо вычислить свободную энергию системы, находящейся в фиксированном объеме
F(T) = E(T) - TS(T) = Sj wj.{ej + kBT. ln(wj)} = Sj wj.{ej + kBT.(-ej/kBT - ln[Z(T)]} = - kBT. ln[Z(T)] | (8.9) |
(в предпоследнем равенстве мы воспользовались формулой 8.5). Затем можно прямо найти энтропию системы: S(T) = —dF/dT (мы с этой формулой уже встречались, но проверьте ее самостоятельно, исходя из определений, даваемых формулами 8.9, 8.6, 8.5 и 8.8), а затем — и ее энергию E(T) º F(T) + TS(T) = F(T) - T(dF/dT).
Существенные, но второстепенные замечания.
1) Внутренняя температура "малой системы" Tin равна температуре термостата Т. Это следует из того, что, согласно приведенным выше определениям и уравнениям, Tin º dE(T)/dS(T) = d[F(T)-T(dF/dT)]/d(-dF/dT) = [dF/dT-T(d2F/dT2)-dF/dT]dT/ (— d2F/dT2)dT = T.
2) Полная энергия складывается из кинетической и потенциальной. Первая зависит только от скоростей частиц, а вторая — только от их места в пространстве, но не от скоростей. "Микросостояние" каждой частицы задается ее координатой в пространстве и ее скоростью. В классической (не квантовой) механике сочетание скоростей и координат может быть любым (и, как мы знаем, квантовое соотношение Гайзенберга, DvDx~![]() /m, — при комнатной температуре — ограничивает сочетание скоростей и координат только у очень легких частиц — электронов). Это значит, что вероятности координат и скоростей можно "расцепить", т. е. w(eкинет+ eкоорд) ~ exp(-eкинет/k BT) x exp(-eкоорд/k BT). Продолжая несложные вычисления (предлагаю их сделать самим!), легко увидеть, что свободные энергии, энергии и энтропии тоже можно "расцепить" на кинетические и координатные части, т. е. F = Fкинет. + Fкоорд. и т. д. Важно, что кинетические части не зависят от конфигурации системы (фактически, они дают только постоянный вклад в теплоемкость) — и их можно отбросить при изучении конформационных превращений. Поэтому в дальнейшем мы будем говорить только о конфигурационных (или конформационных) энергетических спектрах, энергиях, энтропиях и т. д.
/m, — при комнатной температуре — ограничивает сочетание скоростей и координат только у очень легких частиц — электронов). Это значит, что вероятности координат и скоростей можно "расцепить", т. е. w(eкинет+ eкоорд) ~ exp(-eкинет/k BT) x exp(-eкоорд/k BT). Продолжая несложные вычисления (предлагаю их сделать самим!), легко увидеть, что свободные энергии, энергии и энтропии тоже можно "расцепить" на кинетические и координатные части, т. е. F = Fкинет. + Fкоорд. и т. д. Важно, что кинетические части не зависят от конфигурации системы (фактически, они дают только постоянный вклад в теплоемкость) — и их можно отбросить при изучении конформационных превращений. Поэтому в дальнейшем мы будем говорить только о конфигурационных (или конформационных) энергетических спектрах, энергиях, энтропиях и т. д.
3) Выше мы суммировали по микросостояниям — но в рамках классической механики мы можем с тем же успехом интегрировать по координатам и скоростям, задающим микросостояние каждой частицы.
4) Равновесная температура может быть только положительной. Иначе интегрирование вероятностей по скоростям, т. е. ![]() ехр(-mv2/2kT)dv, обращается в бесконечность на больших скоростях — и система "взрывается".
ехр(-mv2/2kT)dv, обращается в бесконечность на больших скоростях — и система "взрывается".
Поэтому стабильное состояние никак не может наблюдаться в тех областях конформационного энергетического спектра, где его плотность (и, значит, энтропия системы) падает с ростом ее энергии: этим областям соответствует Т<0 (см. формулы 8.4 и 8.1 и Рис.8-2а).
5) Величина kBT измеряется в единицах типа "энергия на частицу" или "энергия на моль (=6.02x1023) частиц". Если бы люди с самого начала выражали бы температуру в энергетических единицах, константа Больцмана "kB" не потребовалась бы вообще. Однако история сложилась так, что сначала ввели температурную единицу — "градус" — а потом уже поняли, что "градус" можно всегда пересчитать, конвертировать в "энергию в расчете на частицу": для этого достаточно умножить его (градус) на некоторую константу — "константу Больцмана". Соответственно, размерность kB — "энергия_на_частицу/градус". Величина kB зависит о того, в чем выражается энергия — то ли в "джоуль_на_частицу", то ли в "кал_на_моль частиц". Согласно измерению энергетической величины градуса, kB = 1.38x10-23 (джоуль/частицу)/градус. Однако kB можно выразить и по-иному - в расчете не на штуку, а на моль частиц: 1.38x10-23 (джоуль/частицу)/градус = 1.38x10-23 (джоуль x [6.02x1023]/[6.02x1023 частиц])/градус = [1.38 x 10-23 x 6.02x1023] (джоуль/моль_частиц)/градус = 8.31 (Дж/моль)/градус = 1.99 (кал/моль)/градус. Последнюю величину — 1.99 (кал/моль)/оК — традиционно именуют "газовой постоянной" R.
6) Удельная энтропия часто выражается в энтропийных единицах, "э. ед": э. ед. = (кал/моль)/оК » kB/2. Стало быть, энтропии в 1 э. ед. соответствует е1/2 » 1.65 состояний системы в расчете на частицу.
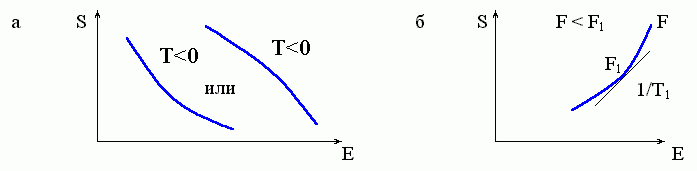
Рис.8-2. Участки графика S(E), которым не может соответствовать никакое стабильное состояние системы. (а) Участки, где энтропия S(E) падает с ростом энергии Е: здесь Т = 1/(dS/dE) < 0. (б) "Вогнутый" участок зависимости S от E: здесь в точке касания (где dS/dE=1/T1) свободная энергия F=E-TS не меньше, а больше, чем на соседних с ней участках кривой S(E); ср. с Рис.8-3.
Теперь, наконец, мы рассмотрим конформационные превращения. Именно для их грамотного рассмотрения мы и повторяли азы статистической физики и термодинамики.
Конформационные превращения бывают постепенные ("континуальные") и резкие ("фазовые"). Попробуем разобраться в их различии, используя графики плотности энергетических состояний — вроде тех, что представлены на Рис.8-1. Для этого прежде всего надо научиться находить на графике стабильное состояние — или состояния — нашей системы при заданной температуре среды.
Пусть нам задано число M(E) состояний нашей системы ("макромолекулы") при каждой ее энергии Е, а также температура Т1 окружающей среды ("термостата"). Как, зная график энтропии S(E) = kB ln[M(E)] и Т1, найти на графике точку, соответствующую стабильному состоянию нашей системы? Температура Т1 показывает, какой наклон dS/dE должен быть в той точке графика, которая нас интересует: здесь dS/dE = 1/T1 — ведь температура у нашей макромолекулы та же, что и у среды.
Но, вообще говоря, таких точек — точек, где график имеет наклон 1/T1 — может быть несколько (см. ниже, Рис.8-5). Какая из них отвечает стабильному состоянию? Рассмотрим касательную к графику в точке Е1, где dS/dE|Е1 = 1/T1. Уравнение такой касательной таково: S-S(E1) = (E-E1)/T1, или: E-T1S = E1-T1S(E1). Величина F1 = E1-T1S(E1) — это просто свободная энергия при температуре T1. Вдоль касательной, как мы видим, величина (E-T1S) неизменна. Всюду слева от касательной величина E - T1S ниже, а всюду справа — выше, чем на касательной (Рис.8-3).
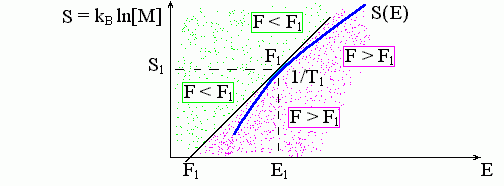
Рис.8-3. Графическое определение свободной энергии. Зависимость энтропии S от энергии E показана жирной кривой S(E). Наклон этой кривой, dS/dE, определяет температуру T. M(E) — плотность спектра энергий состояний системы. F1 — свободная энергия, соответствующая данному энергетическому спектру при температуре Т1. Величина F = E-T1S постоянна вдоль касательной и равна F1. Левее этой касательной — F = E-T1S < F1, правее касательной, — F = E-T1S > F1. Так как кривая S(E) здесь выпукла, т. е. лежит правее и ниже касательной, то точка касания отвечает минимуму свободной энергии при температуре T1. Дальнейшие пояснения — в тексте.
Последнее означает, в частности, что "вогнутым" участкам зависимости S от E (Рис.8-2б) стабильное состояние соответствовать никак не может: здесь в точке касания F не меньше, а больше, чем на соседних с ней участках кривой S(E).
Если кривая S(E) — всюду выпукла, то ее наклон все падает по мере увеличения энергии Е. Тогда каждому наклону 1/Т отвечает только одна точка кривой (Рис.8-1, 8-4), — то есть она, эта точка, и отвечает стабильному состоянию при данной температуре Т. С изменением температуры эта точка постепенно движется, а система — постепенно ("градуально") меняет свое термодинамическое состояние, т. е. энтропию и энергию (Рис.8-4).
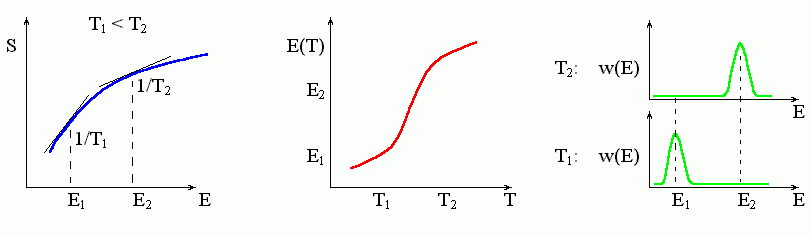
Рис.8-4. Градуальное изменение состояния системы с изменением температуры. Для этого необходимо, чтобы кривая S(E) была выпукла. Зная зависимость S(E) (слева), можно найти зависимости Т(Е) и, далее, Е(Т); последняя показана в центре рисунка. Справа: w(E) — функция распределение молекул по энергиям, т. е. вероятность иметь энергию Е при заданной температуре T; вероятность w(E) пропорциональна exp[-(E-TS(E))/kBT].
Если наклон кривой S(T) то падает, то растет по мере увеличения энергии Е (Рис.8-5), — то касательных с одинаковым наклоном к этой кривой может быть несколько, — и касание самой левой из них (той, у которой F меньше) выделяет стабильное состояние. При этом до какой-то температуры T* "лучшей" будет касательная в районе малых энергий, после T* — "лучшей" будет касательная в районе больших энергий.
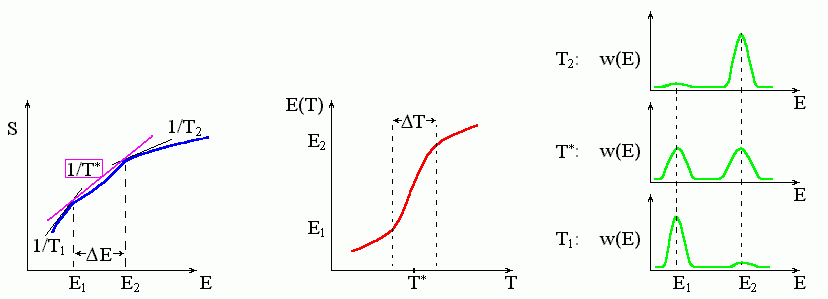
Рис.8-5. Фазовый переход "все-или-ничего" (аналог фазового перехода первого рода в макроскопических телах) характеризуется резким изменением состояния системы при изменении температуры. Для этого необходимо, чтобы кривая S(E) имела бы вогнутый участок: в центре его свободная энергия выше, чем на его краях (см. Рис.8-2б). Переход происходит в узком диапазоне (DT) температур, отвечающем сосуществованию (т. е. примерно равной вероятности) низко - и высокоэнергетичного состояния. Касательные соответствуют: температуре середины перехода T*, а также Т1 < T* и Т2 > T*. Обратите внимание, что температура T* перехода типа "все-или-ничего" (т. е. фазового перехода первого рода) совпадает именно с серединой резкого изменения энергии и с максимальным раздвоением функции распределения по состояниям, w(E).
При температурах, близких к T*, высоко - и низкоэнергетичные структуры будут сосуществовать. И — что наиболее важно — структуры со "средними" энергиями не будут проявляться: из-за "прогиба" кривой соответствующие этим состояниям точки графика лежат справа от касательной, т. е. их свободная энергия — выше, чем у структур, соответствующих точкам касания, а вероятность проявления — ничтожна. Говорят тогда, что два стабильных состояния разделены "свободно-энергетическим барьером". В этих условиях и протекает переход типа "все-или-ничего", который в макроскопических системах называется фазовым переходом первого рода.
Здесь я хочу сразу оценить величину температурного интервала сосуществования высоко - и низкоэнергетичных структур. В середине перехода, при температуре Т*, свободная энергия низкоэнергетичной фазы, F1(T*) = E1-T*S1, равна свободной энергии высокоэнергетичной фазы, F2(T*) = E2-T*S2, т. е. в середине перехода
(E2 — E1) = T*(S2 — S1) . | (8.10) |
При небольшом (на dТ) сдвиге температуры от T* свободные энергии фаз немного меняются, так что разность свободных энергий фаз составляет dF = F1(T*+dT) - F2(T*+dT) = dT (S2 - S1). Фазы сосуществуют (т. е. они примерно равновероятны — скажем, соотношение их вероятностей меняется от 10:1 в пользу первой до 1:10 в пользу второй фазы), пока ехр(-dF/k BT*) лежит между 10 и 1/10, т. е. пока dF/kBT* находится где-то от -2 до +2. В этой области |dT| < 2kBT*/(S2-S1).Значит, температурный интервал сосуществования фаз есть
DT » [2kBT*/(S2-S1)] - [-2kBT*/(S2-S1)] = 4kBT*/(S2-S1) = 4T*[kBT*/(E2-E1)] | (8.11) |
Рассмотрим поучительный численный пример. Если T*~300оК (т. е. kBT*~0.6 ккал/моль), а E2-E1 составляет, как то характерно для плавления белков, порядка 50 ккал/моль [т. е. 50/(6x1023) ~ 10-22 килокалорий_на_штуку], то DT — порядка десяти градусов. Если же E2-E1 составляет порядка 50 килокалорий_на_штуку (как при плавлении льдины размером с бутылку), то DT — порядка 10-23 градуса.
Иными словами, для фазовых переходов типа "все-или-ничего" в малых системах и — в гораздо большей степени — для фазовых переходов первого рода в макроскопических системах, — характерен резкий скачок энергии в узком диапазоне температур. В макроскопических системах этот диапазон практически бесконечно узок, а для макромолекул он охватывает несколько градусов, т. е. тоже узок по сравнению с "диапазоном наблюдения", обычно охватывающим от 0оС до 100оС. А вот в небольших олигопептидах резкого скачка нет — здесь диапазон изменения энергии может охватывать все "экспериментальное окошко" исследуемых температур.
Несколько слов о фазовых переходах второго рода. Если для переходов первого рода характерен скачок энергии системы (и ее энтропии, объема и плотности), то для переходов второго рода характерен лишь излом зависимости скорости изменения энергии с температурой, — иными словами, скачок теплоемкости.
Подчеркиваю, что переход второго рода происходит в точке излома (Рис.8-6), а вовсе не посередине часто идущей за ним более или менее "S-образной" зависимости энергии (или другого наблюдаемого параметра) от температуры.
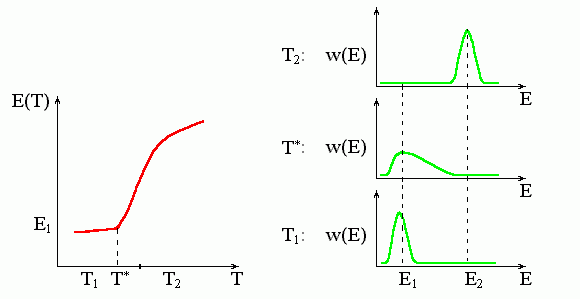
Рис.8-6. Характерная зависимость энергии, E(T), и функции распределения, w(E), при фазовом переходе второго рода. T* — температура этого фазового перехода. При этой температуре функция распределения молекул по энергиям, w(E), резко уширяется, и энергия начинает (или кончает — смотря откуда смотреть) резко изменяться. Обратите внимание, что температура фазового перехода второго рода T* совпадает именно с началом (а не с серединой) резкого изменения энергии.
На рисунках 8-4 — 8-6 я специально представил случаи, когда энергия системы сначала меняется медленно с изменением температуры, потом — резко, а потом — снова медленно. Как вы видите, такое "S-образное" поведение Е(Т) совместимо и с фазовым переходом первого рода, и с градуальным изменением системы при отсутствии какого-либо фазового перехода вообще, и с градуальным изменением, начало которому кладет фазовый переход второго рода.
Я прошу обратить на это особое внимание, так как среди не-физиков существуют два непонятно откуда взявшихся предрассудка: что "переход" соответствует именно середине всякого S-образного изменения, и что если этот "переход" не первого рода — значит второго. Это не так!
Обратите внимание: хотя кривые Е(Т) на рисунках 8-4 — 8-6 выглядят, в общем, одинаково, — поведение кривых распределения по энергиям, w(Е), при переходе первого рода (т. е. типа "все-или - ничего") выглядит совсем не так, как при всех остальных переходах. Для переходов типа "все-или-ничего" — и только для них — характерно наличие двух пиков у w(Е), т. е. сосуществование двух фаз. Поэтому для выяснения того, как — прыжком или постепенно — происходит переход между двумя крайними состояниями, — недостаточно увидеть, что энергия или какой-то наблюдаемый параметр резко меняется в узком диапазоне температур. Здесь нужны дополнительные измерения, которые мы рассмотрим в одной из дальнейших лекций.
В заключение поговорим о кинетике конформационных превращений. Точнее — о том, почему некоторые превращения идут очень медленно. Что здесь значит "медленно"? Предположим, вы знаете время элементарного шага по ходу процесса. Например: один остаток включается во вторичную структуру за 1 наносекунду (нс). Вы знаете также, что в цепи — 1000 звеньев. А процесс идет не за 1000 нс, а за 1000 секунд. Это и есть "медленно": на порядки медленнее, чем можно ожидать, зная время элементарного шага и число таких шагов. Надо понять — почему...
"Медленность" процессов порой связана с медленностью диффузии при высокой вязкости. Однако часто — подчеркиваю, не всегда, но часто — медленность процесса связана с преодолением высокого свободно-энергетического барьера. Особенно это типично для переходов типа "все или ничего", где свободно-энергетический барьер разделяет две фазы (Рис.8-5); здесь он всегда значителен. Такой барьер очень похож на активационный барьер в химических реакциях, — только в данном случае он имеет и энергетическую, и энтропийную составляющие. Напомню, как оценивается скорость такой "барьерной" реакции согласно классической теории переходных состояний.
Рассмотрим простейший процесс, по ходу которого система переходит из состояния А в состояние В. Пусть на пути процесса "А®В" есть один "барьер" # (Рис.8-7), и ни на этом пути, ни "сбоку" от него нет никаких "ловушек" (Рис.8-8: состояний Л, более стабильных, чем исходное A, но менее стабильных, чем конечное B; наличие ловушек усложняет процесс, но не меняет его принципиально).
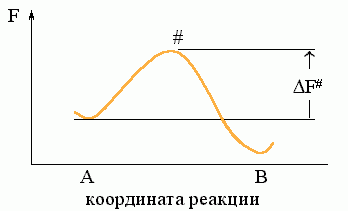
Рис.8-7. Преодоление свободно-энергетического ("активационного") барьера # при переходе из состояния "А" в состояние "В". DF# — свободная энергия барьера (перехoдного состояния).
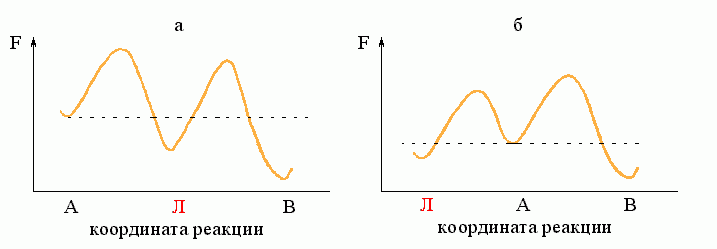
Рис.8-8. "Ловушка": ею может быть как интермедиат "Л" (а), так и побочное (для реакции А®В) состояние "Л" (б). Для наличия кинетической "ловушки" важно только, что она более стабильна, чем исходное состояние "А", но менее стабильна, чем конечное состояние "В", и что на пути из "Л" в "В" находится более высокий свободно-энергетический барьер, чем на пути из А в Л.
Тогда скорость процесса определяется (1) населенностью барьерного ("перех![]() дного") состояния, и (2) скоростью перехода из барьерного в "за-барьерное" состояние.
дного") состояния, и (2) скоростью перехода из барьерного в "за-барьерное" состояние.
Если DF# — свободная энергия барьера, отсчитанная от свободной энергии начального состояния, причем DF# >> kBT, и "ловушек" (Рис.8-8) нет, а в начальном состоянии находится n "частиц" (синонимы: молекул, систем и т. д.), — то на барьере, в каждый момент времени, находится n# ~ n. exp(-DF#/kBT) частиц. Пусть каждая из этих на-барьерных частиц переходит за барьер за время t (время элементарного шага реакции). Тогда за время порядка t за барьер перейдут все n# на-барьерных частиц. На то, чтобы за барьер перевалили бы все n частиц — понадобится n/n# шагов, т. е. время порядка
t ~ t (n/n#) ~ t exp(+DF#/kBT) | (8.12) |
При наличии "ловушки", Л, таким же образом оценивается время перехода из А в Л и из Л в В.
Уточнение. Если на пути из А в В находится не одно, а s последовательных промежуточных состояний с со свободной энергией, близкой к DF#, — то скорость процесса замедляется в s2 раз по сравнению с оценкой (8.12). Это замедление связано с диффузией частиц по верхушке барьера. Однако обычно этот диффузионный фактор не играет существенной роли.
Лекция 9
Обсудим теперь, зная базовую физику, стабильности вторичной структуры и кинетику ее образования.
Начнем с a-спиралей. Первая водородная связь в ней, (СО)0 — (HN)4, фиксирует конформации трех остатков — 1, 2, 3; следующая водородная связь, (СО)1 — (HN)5, дополнительно фиксирует конформацию только одного остатка — остатка 4; связь (СО)2 — (HN)6 дополнительно фиксирует остаток 5, и т. д.
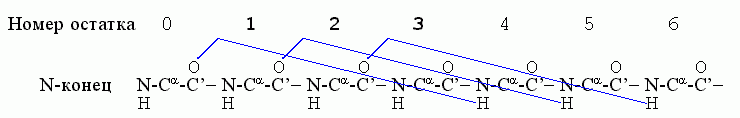
Значит, если в спирали фиксировано n остатков, то их фиксирует n-2 водородные связи. Рассмотрим свободную энергию образования такой спирали из клубка в водном окружении ("клубок" — это полимер без структуры и без взаимодействия дальних по цепи звеньев). Эту свободную энергию можно записать как
DFa = Fa — Fклуб. = (n-2)fH — nTSa = -2fH + n(fH —TSa) | (9.1) |
Здесь fH — свободная энергия водородной связи в a-спирали (вы помните, что fH — не просто энергия, как было бы в вакууме: в нее входит и энергия, и энтропия перестроек водородных связей в водном окружении), а Sa — потеря энтропии при фиксации одного остатка в спирали.
Вы видите, что в DFa есть два члена. Один (-2fH) не зависит от длины спирали; величина
fINIT = -2fH | (9.2) |
традиционно называется свободной энергией инициации спирали (на самом деле, fINIT — суммарная свободная энергия обеих границ спирали с клубком — учитывает и инициацию, и терминацию спирали). Другой член, n(fH-TSa), прямо пропорционален длине спирали; величина
fEL = (fH-TSa) | (9.3) |
называется свободной энергией элонгации спирали на один остаток. В общем виде,
DFa = fINIT + n.fEL. | (9.4) |
При этом отношение вероятности чисто спирального состояния цепи из n остатков к ее же чисто клубковому (и начисто лишенному спиральных примесей) состоянию, равно
exp(-DFa/kT) = exp(-fINIT/kT).[exp(-fEL/kT)]n = ssn | (9.5) |
Здесь я использовал общепринятые обозначения:
— фактор элонгации спирали: s = exp(-fEL/kT);
— фактор инициации спирали: s = exp(-fINIT/kT);
ясно, что s<<1, так как s = exp(-fINIT/kT) = exp(+2fH/kT); а свободная энергия водородной связи — большая отрицательная величина, порядка нескольких kT.
Прежде, чем говорить о том, как экспериментально определяются величины s и s, — выясним общий вопрос о том, как образуется спираль при изменении условий среды (температуры, растворителя и т. д.) — переходом "все-или-ничего" или постепенно?
Поначалу кажется, что такая отличная от клубка структура, как спираль, должна "вымораживаться" из него путем фазового (т. е. "все-или-ничего") перехода — как лед из воды...
Однако на этот счет есть теорема Ландау, которая гласит, что в системе, где обе фазы одномерны, — фазовый переход первого рода невозможен. Попытаюсь объяснить эту теорему.
Прежде всего — что значит "одномерность". Это значит, что размер границы, "стыка" фаз не зависит от размера кусков этих фаз. В этом смысле и спираль, и клубок в полимере — одномерны. Рисунок 9-1 поясняет, что граница (стык) спирали и клубка не зависит ни от длины спирали, ни от длины клубка, — в то время как поверхность, граница трехмерной фазы (например, льдинки в воде) зависит от ее размера. Соответственно, свободная энергия границы спирали и клубка не зависит от их длин, а свободная энергия поверхности трехмерной фазы растет как n2/3 с ростом числа n вовлеченных в нее частиц.
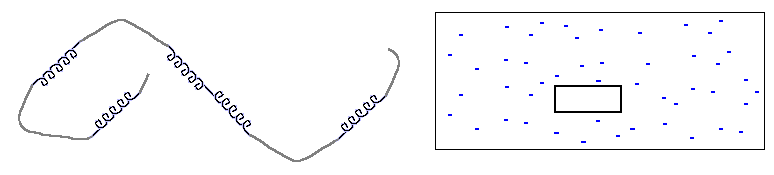
Рис.9-1. Сравнение одномерной (клубок со спиралями) и трехмерной (льдинка в воде) систем. Размер стыка спирали и клубка не зависит их длин; поверхность трехмерной льдинки зависит от ее размера.
Теперь — что значит "происходит как фазовый переход первого рода". Это значит, что при температуре перехода стабильной может быть либо одна, либо другая фаза, но перемешивание фаз (например, льда и воды) ведет к повышению свободной энергии, и потому нестабильно. Здесь вас не должна вводить в искус картина плавающего в воде льда: при любой температуре такое состояние неустойчиво (из-за повышенной свободной энергии на границе воды со льдом), и со временем лед или растает, или охватит всю воду — конечно, если нет тока подземного тепла, течений и других неравновесных вещей...
Выгодно ли сосуществование фаз в трехмерной системе? Нет. Почему? Обратимся опять к Рис.9-1. Рассмотрим температуру, где безграничная вода и безграничный лед имеют равную свободную энергию (это и есть условие "точки перехода"). Если в воде плавает льдина из n молекул, свободная энергия границы пропорциональна xn2/3, где n2/3 — характерное число пограничных молекул, а x>0 — энергия границы в расчете на одну из них. (Замечание. Если x<0, "перемешивание" — оно при этом всегда термодинамически выгодно — произойдет на молекулярных масштабах, и двух фаз не будет вообще). Значит, поверхность льдинки повышает свободную энергию на xn2/3. Правда, льдинка имеет еще позиционную энтропию, так как может находиться в разных точках сосуда. Но эта энтропия не превосходит величину порядка k. ln(N), если в сосуде — всего N молекул (т. е. N точек, с которых может начинаться льдинка). Итого — свободная энергия льдинки — порядка [xn2/3 - kТ. ln(N)]. Но логарифм — очень "слаборастущая" при больших N функция. Если льдинка занимает заметную часть сосуда (скажем, n~N/10), и N очень велико [скажем,], то ln(N) [в данном случае - 23] очень мал по сравнению с (N/10)2/3 [в данном случае - с 1 ], — то есть в свободной энергии льдинки доминирует граничный член xn2/3, а он противится ее образованию... Поэтому в трехмерной системе макроскопические фазы разделяются ("льдинки" из нескольких молекул — не в счет: это просто микроскопические, локальные флуктуации), и фазовый переход первого рода в ней возможен.
А выгодно ли сосуществование фаз в одномерной системе? Оказывается, да. Рассмотрим опять температуру "середины перехода", где спираль и клубок имеют равную свободную энергию, т. е. где fEL=0. Свободная энергия обеих границ спирали и клубка, fINIT, не зависит ни от размера спирали, ни от размера клубка. Позиционная энтропия спирали длины n в цепи длины N равна k. ln(N-n). Итого, свободная энергия этой плавающей спирали есть fINIT - kТ. ln(N-n). При больших N, член с ln(N-n) всегда доминирует в этом выражении, даже если n ~ 0.9 N; а этот логарифмический член понижает свободную энергию и способствует внедрению спирали в клубок (и, точно так же, — клубка в спираль)... Поэтому в одномерной системе фазы не разделяются, они стремятся перемешаться, — а раз так, то и фазовый переход первого рода (или типа "все-или-ничего") невозможен — при достаточно большой длине цепи. Теорема Ландау доказана.
Теперь можно поставить вопрос — при каких характерных длинах цепи начинается смешение клубковой и спиральной фаз?
Рассмотрим цепь из N звеньев при температуре "середины перехода", где спираль и клубок имеют равную свободную энергию, т. е. fEL=0. При этом свободная энергия элонгации спирали (а равно и клубка) — ноль, ее инициации — fINIT, а число возможных положений спирали в цепи из N звеньев — порядка N2/2 (она может начинаться и кончаться в любом месте при единственном условии, что ее длина — не менее трех остатков); и ни расположение спирали в цепи, ни ее длина (при fEL=0) не влияют на ее свободную энергию. Для получения качественной оценки — пренебрежем мелочами (цифрами) по сравнению с главным (буквами). Итак: размещений спирали — порядка N2, т. е. их энтропия — k x 2ln(N), а полная свободная энергия внедрения куска новой фазы (спирали с флуктуирующими концами) в цепь длины N — примерно fINIT - 2kТ. ln(N). Если она, эта свободная энергия, больше нуля — новая фаза не внедрится; если она меньше нуля — новая фаза может внедриться неоднократно. Значит, смешение клубковой и спиральной фаз начинается в кусках длины N ~ n0, а величина n0 получается из уравнения fINIT - 2kТ. ln(n0) = 0. Итак: характерная длина кусков спирали и клубка в середине перехода
n0 = exp(+fINIT/2kТ) = s - 1/2 . | (9.6) |
Точка (температура) середины перехода на опыте находится как та точка, где спиральность очень длинного полипептида составляет 50% (спиральность обычно измеряют при помощи КД спектров, как о том говорилось выше; при 50% спиральности КД спектр полипептида выглядит как полусумма спектров клубкообразного полипептида и полипептида, спирального на 100%). В этой точке fEL=0, т. е. s = exp(-fEL/kT) = 1.
Измеряя в тех же условиях (когда s=1) зависимость спиральности от длины полипептида, можно найти n0, — как ту длину цепи, при которой ее средняя степень спиральности в 4 раза меньше, чем у очень длинной цепи (т. е. чем 50%).
Точный расчет и доказательство того, что цепь из n0 = s -1/2 звеньев имеет именно (1/4) 50% = 12% спиральности при s=1, — выходит за рамки нашего курса. Однако получить приблизительную оценку нетрудно. В самом деле, в цепи из n0 звеньев сумма вероятностей всех состояний, включающих a-спираль, близка — по определению — к 50%. А так как состояние "со спиралью" обычно имеет вид типа "клубок_на_одном_конце_цепи — спираль — клубок_на_другом_конце_цепи", то, в среднем, эта спираль — если она есть вообще (а вероятность этого "вообще" — 50%) — включает 1/3 звеньев цепи. Итого — средняя спиральность цепи из n0 звеньев приближенно равна 50% x (1/3) = 17%.
Наконец, зная n0, — можно вычислить fINIT и s. Для большинства аминокислот n0 » 30, fINIT » 4 ккал/моль, и s » 0.001.
Теперь мы можем найти свободную энергию образования водородной связи (вкупе со всеми сопутствующими образованию Р-связи в a-спирали взаимодействиями): согласно (9.2), fH = - fINIT/2 » -2 ккал/моль. Можно найти и конформационную энтропию, теряемую при фиксации одного звена в a-спирали: согласно формуле (9.3), TSa = fH » 2 ккал/моль.
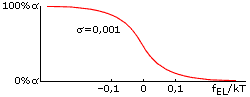
Оба параметра стабильности спирали — и fEL, и fINIT — зависят от температуры, но по-настоящему сильно влияет на стабильность спирали именно отклонение величины fEL от 0. Дело в том, что в спирали, состоящей из ~n0 звеньев, это отклонение умножается на большое число n0 — и уже в таком виде входит в свободную энергию спирали. Когда величина fELxn0/kT составляет порядка +1 (более точная оценка: fELxn0/kT = +2), спиральность практически исчезает, а когда fELxn0/kT = -2, — практически исчезает клубок. Значит, при n0»30, переход спираль-клубок в очень длинных (N>>n0) полипептидных цепях происходит в области -0.07 < fEL/kT < 0.07 (Рис.9-2). Это — пример резкого, кооперативного, — но не фазового (т. к. ширина его не стремится к нулю по мере роста длины цепи) перехода.

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 |
