
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Видно, что только спираль aR (a-правая) лежит достаточно глубоко внутри области, разрешенной для аланина (и для всех других остатков). Другие спирали лежат либо на краю этой области (например, левая спираль aL или правая спираль 310), где конформационные напряжения уже возрастают, либо в области, доступной только глицину.
Поэтому можно ожидать, что именно правая a-спираль должна быть, как правило, более стабильной, и потому преобладать в белках — что и наблюдается. В правой a-спирали (Рис.7-5) все атомы упакованы оптимально: плотно, но без напряжений; поэтому не удивительно, что в белках таких спиралей много, а в фибриллярных белках они достигают гигантской длины и включают сотни аминокислотных остатков.

Рис.7-5. Правая a-спираль. (а) Атомарная структура. R — боковые группы. Голубые линии — водородные связи. (б) Схематическое изображение одного витка той же a-спирали, вид с торца. Стрелка показывает поворот спирали (в расчете на один остаток) по мере ее приближения к нам. Картинка (а) взята из [3] и адаптирована.
Левых a-спиралей в белках практически нет. Нет и спиралей 27, которые, мало того что лежат на самом краю разрешенной области, но еще имеют энергетически невыгодный, почти прямой угол схождения N-H и О=С групп.
Практически нет в белках и спиралей p. Они тоже лежат на самом краю разрешенной области, да еще и витки в них слишком широки, так что p-спирали имеют энергетически невыгодную пустую "дырку" на оси. А вот спирали 310 (в основном — правые, — левые пригодны практически лишь для глицинов) в белках есть — правда, в виде коротких (из трех-четырех остатков) и деформированных фрагментов (из-за стерических напряжений: слишком тугая спираль! — соответствующая ей конформация лежит на самом краю разрешенной области).
Отметим одно свойство спиралей, хорошо видное на Рис.7-5а: на их N-конце сидят свободные от внутриспиральных водородных связей Н атомы N-H групп, а на С-конце — свободные от водородных связей О атомы С=О групп. Так как электронное облако с Н атома частично стянуто электроотрицательным N атомом, а электроотрицательный О атом сам стягивает электрон с С' атома, — N-конец спирали несет положительный, а С-конец — отрицательный парциальный заряд. То есть спираль представляет собой длинный диполь: величина суммарного (поставляемого тремя NH-группами) парциального "+" заряда на ее N-конце составляет около половины протонного, а "-" заряда на С-конце a-спирали — около половины электронного заряда.
Теперь рассмотрим регулярные структуры без водородных связей внутри каждой, но соединенные водородными связями между собой.
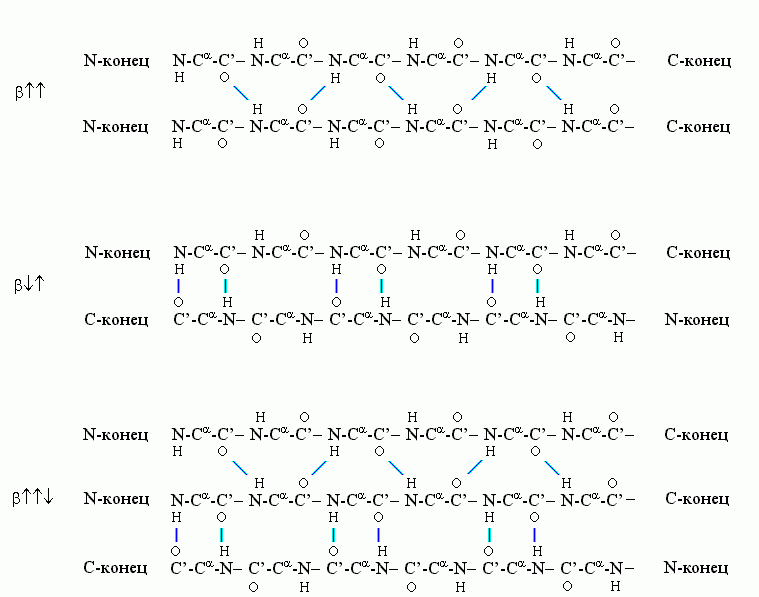
Рис.7-6. Схема хода цепи и расположения водородных связей в параллельной b![]()
![]() , антипараллельной b
, антипараллельной b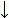
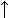 и смешанной b
и смешанной b![]()
![]()
![]() b-структуре. Видно, что Н-связи одного остатка в b-тяже направлены одну сторону, следующего — в противоположную, и т. д.
b-структуре. Видно, что Н-связи одного остатка в b-тяже направлены одну сторону, следующего — в противоположную, и т. д.
Почти вытянутые (все углы в главной цепи — почти trans), слегка скрученные цепи образуют b-структуру. Она бывает (Рис.7-6) параллельной (b![]()
![]() ), антипараллельной (b) и смешанной (состоящей из b
), антипараллельной (b) и смешанной (состоящей из b![]()
![]() и b
и b![]()
![]() ).
).
b-структура стянута водородными связями (символы / , \ и | на схемах 7-6). Она существует в виде более или менее крупных листов. Так как поверхность b структуры — рифленая, ее еще называют "складчатой b-структурой" (Рис.7-7).
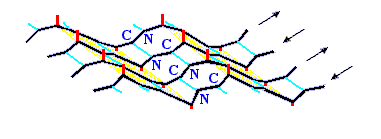
Рис.7-7. Лист b структуры имеет складчатую поверхность. Боковые группы (см. маленькие отростки) расположены на складках; каждая обращена в ту же, что и складка, сторону, т. е. направленные вниз и вверх боковые группы чередуются вдоль b-тяжа. Картинка взята из [3] и адаптирована.
b-структурные листы всегда несколько скручены (Рис.7-8а) из-за того, что несколько скручены (Рис.7-8б) отдельные b-тяжи — и потому по ходу тяжа несколько меняется направленность водородных связей. А тяжи, в свою очередь, скручены из-за того, что наиболее энергетически выгодная конформация остатков с боковыми группами сдвинута к центру стерически разрешенной области (Рис.7-8в). Скрученность отдельного b-тяжа — левая (у L аминокислот! — у D было бы наоборот): вы видите (Рис.7-8б), что боковые группы тяжа поворачиваются по часовой стрелке (на »-165o на каждый остаток) по мере приближения тяжа к нам.
В результате, поворачиваются и Н-связи (на »-165o на каждый остаток, т. е. на -330o=+30o на пару остатков - повторяющуюся единицу b-структуры), так что угол между соседними тяжами b-структуры (если смотреть с кромки листа, Рис.7-8а) обычно составляет около -25о ("минус", как всегда, означает, что ближний к нам b-тяж повернут по часовой стрелки относительно более далекого). Таким образом, b-лист имеет левое скручивание, если смотреть с края этого листа (и правое, если смотреть вдоль b-тяжей на поворот линии Н-связей).
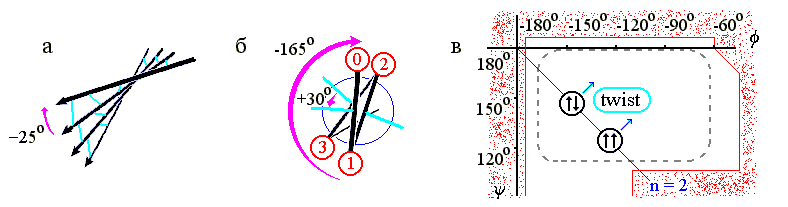
Рис.7-8. (а) Скрученность b-листа. b-тяжи изображены стрелками, водородные связи между ними — голубыми линиями. (б) Схематическое изображение одного витка b-тяжа, вид с торца. Кружки — боковые группы; их номера возрастают по мере удаления от читателя. Голубые линии укаывают направление С=О групп, завязывающих Н-связи в листе, большей стрелкой — поворот b-тяжа при приближении к нам на один остаток, меньшей стрелкой — поворот направленных в одну сторону водородных связей при приближении к нам на два остатка. (в) Конформация идеальной (не скрученной) параллельной (![]()
![]() ) и антипараллельной (
) и антипараллельной (![]()
![]() ) b-структуры для поли(Gly), и усредненная конформация реальной (сложенной из L аминокислот) скрученной (twist) b-структуры. Пунктир показывает область энергетического минимума для отдельно взятого остатка Ala; контуром показаны границы области разрешенных конформаций этого остатка. Диагональ jy-карты соответствует плоской периодичной структуре, имеющей 2 остатка на виток. Над диагональю лежат левые (L) спирали, под ней — правые (R). Картинки (а), (в) взяты из [3] и адаптированы.
) b-структуры для поли(Gly), и усредненная конформация реальной (сложенной из L аминокислот) скрученной (twist) b-структуры. Пунктир показывает область энергетического минимума для отдельно взятого остатка Ala; контуром показаны границы области разрешенных конформаций этого остатка. Диагональ jy-карты соответствует плоской периодичной структуре, имеющей 2 остатка на виток. Над диагональю лежат левые (L) спирали, под ней — правые (R). Картинки (а), (в) взяты из [3] и адаптированы.
Есть спирали и без водородных связей, где плотная (а значит — энергетически выгодная) упаковка держится чисто на Вандерваальсовых контактах. Это — полипролиновая спираль. При этом три скрученные в довольно растянутую левую спираль цепи образуют правую суперсуперспираль — они плотно закручиваются друг вокруг друга. Из двух возможных типов полипролиновой спирали для нас важна спираль poly(Pro)II: она реализуется в коллагене. В этой спирали пептидные группы пролинов находятся в обычной (trans) конформации. Отложим более подробное рассмотрение коллагеновой спирали до соответствующего места курса, а пока ограничимся общим ее видом (Рис.7-9) и отметим на Рис.7-4 область соответствующей ей конформации цепи: видно, что она довольно близка к b структуре.
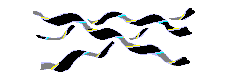
Рис.7-9. Общий вид тройной правой суперспирали из левых спиралей Poly(Pro)II.
Параметры наиболее важных регулярных вторичных структур белковых цепей суммируются в следующей таблице:
Таблица 7/1. Основные геометрические параметры наиболее распространенных в белках вторичных структур.
Структура | H-связь | Остаток/виток | Смещение/остаток ( | f | y |
Спираль aR | CO0—HN+4 | +3.6 | 1.5 | -600 | -450 |
Спираль (310)R | CO0—HN+3 | +3 | 2.0 | -500 | -250 |
Лист b | меж цепей* | -2.3 | 3.4 | -1350 | +1500 |
Лист b | меж цепей* | -2.3 | 3.2 | -1200 | +1350 |
Спираль Poly(Pro) II | нет | -3 | 3.0 | -800 | +1550 |
* Расстояние между тяжами в b-листе: 4.8![]()
Примечание. Данные взяты из [3, 6]. Все цифры округлены. "+" означает правую спираль, "-" — левую.
Кроме регулярных, в полипептидных цепях есть еще и нерегулярные вторичные структуры, — т. е. стандартные структуры, не образующие длинных периодических систем.
Это — так называемые b-изгибы ("b" — потому, что они часто стягивают верхушки соседних b-тяжей в антипараллельных b шпильках). Характерный вид наиболее важных b-изгибов и конформации входящих в них остатков представлены на Рис.7-10. Сравните Рис.7-10в с Рис.7-4 и Таблицей 7/1, и обратите внимание на то, что изгибы I (и особенно III) близки по конформации к витку спирали 310.
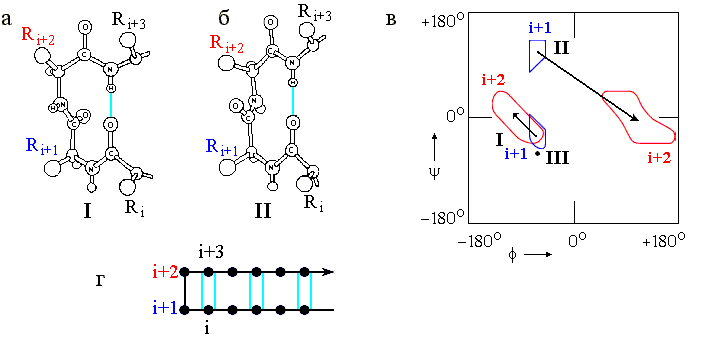
Рис.7-10. b-изгибы. (а) b-изгиб типа I (b-изгиб типа III очень на него похож и потому не нарисован отдельно). (б) b-изгиб типа II. Его основное отличие от b-изгиба I — переворот пептидной группы, соединяющей остатки i+1 и i+2. (в) Конформации фиксируемых водородной связью остатков i+1 и i+2 в b-изгибах. В b-изгибе III оба остатка i+1 и i+2 имеют одинаковую конформацию (отмечена жирной точкой). Конформации остатков i и i+3 в b-изгибах не фиксированы; они фиксируются b-структурой - если она прирастает из изгиба, как на рисунке (г), где дана схема b-шпильки с b-изгибом в ее вершине. Картинки (а), (б), (в) взяты из [3] и адаптированы.
В заключение — несколько слов о том, как экспериментально обнаруживается вторичная структура.
Конечно, если сделан рентген (или точный многомерный ЯМР) белка — вторичная структура берется из атомных координат.
Впрочем, ЯМР (ядерный магнитный резонанс), который хорошо фиксирует сближенность (до 4 — 5 и менее ![]() ) ядер Н-атомов, позволяет определять вторичную структуру даже тогда, когда полную атомную структуру белка построить еще не удается.
) ядер Н-атомов, позволяет определять вторичную структуру даже тогда, когда полную атомную структуру белка построить еще не удается.
Метод ЯМР основан на возбуждении радиоволнами ориентированных в сильном магнитном поле ядер, — тех ядер, которые имеют нечетное число нуклонов (протонов и нейтронов): только они имеют спин и, вследствие этого, — магнитный момент. В белке это — природные "легкие" водороды (1H), а также вводимые изотопы (13С, 15N и т. д.). Магнитный резонанс наступает на радиочастоте, характерной для данного атома, причем эта частота слегка модифицируется его соседями по химическим связям и по пространству (что и позволяет судить, атом какого остатка возбудился). Возбужденное ядро может передать свое возбуждение соседнему с ним в пространстве ядру с магнитным моментом, и оно отрапортует о полученном возбуждении уже на своей частоте (что и позволит судить о сближенности этих двух магнитных ядер).
Для a-спиралей особенно характерна сближенность H-атома группы CaH с H-атомом NH-группы 4-го от нее (к С-концу цепи) остатка, а для b-структуры — сближенность Н-атомов NH - и CaH-групп у остатков, непосредственно соседствующих по цепи, и у остатков, связанных Н-связями в b-листе (Рис.7-11).
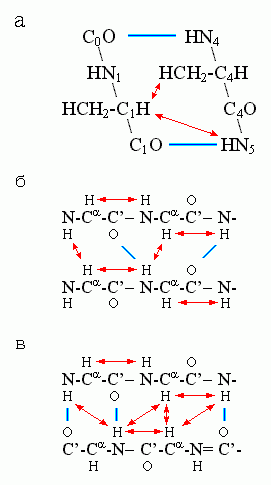
Рис.7-11. Сближенность («) ядер водородных атомов, наиболее характерная для a-спирали (а), параллельной (б) и антипараллельной (в) b-структуры. Индексы при атомах главной цепи в рисунке (а) показывают взаимное расположение остатков в цепи.
Однако наиболее важную, пожалуй, роль в определении вторичной структуры играет метод кругового дихроизма (КД). Он не требует знания общей пространственной структуры белка. Наоборот, структурное исследование белка обычно начинается с получения спектров КД. Метод КД основан на различии в поглощении право - и левополяризованного света в спиралях различной закрученности. Из-за этого различия в поглощении плоскополяризованный свет превращается в эллиптически поляризованный.
Характерные спектры эллиптичности в области "дальнего" ультрафиолета (190-240 нм) приведены на Рис.7-12. Показанные спектры зависят от асимметрии окружения пептидных групп и потому рапортуют о том, есть ли в белке вторичная структуре, какая, и сколько ее.
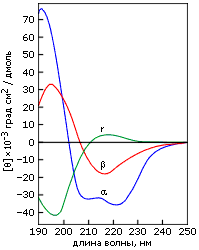
Рис.7-12. Характерные формы спектров КД для полилизина в форме a-спирали (a), b-структуры (b) и неупорядоченного клубка (r). Картинка взята из [6] и адаптирована.
Пептидные группы оптически возбуждаются в "дальнем УФ", при длине волны порядка 200 нм. Это — примерно вдвое большая длина волны, чем та, на которой возбуждаются отдельные атомы. Причина того, что пептидная группа возбуждается более длинноволновым (т. е. менее "жестким") светом, — в делокализации электронов пептидной группы по нескольким атомам, о чем мы уже говорили.
Еще больше делокализованы электроны в ароматических группах — там они "размазаны" не по трем, как в пептидной группе, а по шести атомам. Спектры КД ароматических групп приходятся на длину волны ~250-280 нм (хотя "хвост" этих спектров доходит до ~220 нм). В этом диапазоне длин волн, ~250-280 нм, (в "ближнем" ультрафиолете) изучают асимметрию окружения ароматических боковых групп, — т. е. эффекты, связанные с образованием уже не вторичной, а третичной структуры белка.
В скобках отмечу, что при еще большей делокализации электрона (в более крупных молекулах с кратными связями) — он начинает возбуждаться уже не ультрафиолетовым, а видимым светом (4нм): свечение таких молекул видно на глаз, т. е. они являются красителями.
Кроме ультрафиолетовых спектров, для регистрации вторичной структуры полипептидов и белков используются инфракрасные спектры. Они отражают различия в колебаниях пептидных групп, вовлеченных и не вовлеченных в разные вторичные структуры (Рис.7-13). Эти измерения более сложны, чем измерения УФ-спектров, так как обычная вода (H2O) поглощает в той же области; поэтому такие измерения обычно проводятся в тяжелой воде (D2O). Кроме того, они требуют больше белка, чем измерения УФ-спектров, и более высоких концентраций белка в растворе.
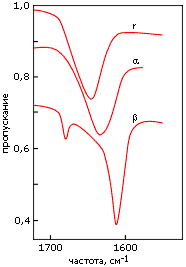
Рис.7-13. Характерные формы инфракрасных спектров пропускания, измеренных в тяжелой воде (D2O) для полилизина в форме a-спирали (a), b-структуры (b) и неупорядоченного клубка (r). Измерения, в данном случае, проводились в области "амид I", отражающей колебания С=О связи. Картинка взята из [6] и адаптирована.
Лекция 8
Теперь можно было бы перейти к рассказу об образовании и распаде вторичной структуры.
Однако прежде я хочу поговорить об основах статистической физики и термодинамики "вообще", т. к. без этого трудно рассказывать о и стабильности вторичной структуры, и о стабильности белков, и о кооперативных переходах в полипептидах и белках, и о кинетике этих переходов.
Термодинамика дает представление о типах возможных кооперативных переходов в системах, состоящих из очень большого числа частиц. Статистическая физика позволяет указать, когда и какие переходы произойдут в рассматриваемой системе и описать детали этих переходов, исходя из свойств рассматриваемых частиц и взаимодействий между ними.
Прежде всего мы рассмотрим основные понятия статистической физики и термодинамики — энтропию, температуру, свободную энергию и статистическую сумму.
Итак, системы с большим числом степеней свободы (т. е. состоящие из большого числа молекул — или даже из одной большой и гибкой молекулы) описываются при помощи статистической физики. "Статистической" — ибо число конфигураций таких больших систем колоссально. Только один пример: если каждое из N звеньев цепи может находиться всего в двух возможных конфигурациях (например: "спиральном" и "вытянутом"), то вся N-звенная цепь имеет 2N возможных конфигураций. То есть "нормальная" для белка цепь из 100 звеньев может иметь, по крайней мере, 2100, или около 10,000,000,000,000,000,000,000,000,000,000 конфигураций. Это — очень много. А ведь на опыте в пробирке находятся миллиарды таких цепей, — не говоря о растворителе! И если бы мы захотели инвентаризовать все конфигурации этих цепей, — мы бы пропали навсегда. Но нас, конечно, будут интересовать более простые и разумные вещи, например, — средняя (т. е. статистически усредненная) спиральность цепей и то, как эта спиральность меняется с нагреванием. А замечательным свойством статистического, т. е. пренебрегающего всеми несущественными подробностям усреднения является кардинальное упрощение ситуации.
Важнейшую роль в статистическом усреднении играет энтропия. Она говорит, сколько конфигураций системы (или, как говорят, сколько ее микроскопических состояний) соответствует наблюдаемому нами, т. е. макроскопическому (усредненному длительностью нашего наблюдения и большим числом одновременно наблюдаемых молекул) состоянию изучаемой системы. Мы уже говорили на эту тему, — но тогда мы рассматривали, по существу, только один частный случай: тогда число "микроскопических состояний" определялось объемом, в который была заключена молекула, а ее энтропия была пропорциональна логарифму этого объема (причем нахождение в данном объеме — например, в данной комнате — и было "макроскопическим состоянием" молекулы).
Здесь уместно ответить на естественный вопрос: почему физики предпочитают рассматривать логарифм числа микросостояний, а не само это число? Дело в том, что, если рассматривать много отдельных систем (например, — отдельных молекул), то мы увидим, что их объемы, энергии, числа степеней свободы при этом складываются, а количества возможных состояний — перемножаются. Это различие неудобно, оно сделало бы громоздким все расчеты. Но вот логарифмы чисел возможных состояний складываются при перемножении этих чисел [вы помните, что ln(AB)=ln(A)+ln(B)], — складываются, как энергии или объемы. Это однотипность удобна при решении задач, и к тому же операция сложения позволяет легко воспользоваться всей мощью дифференциального исчисления.
Теперь — температура. Она тесно связана с энтропией: нет большого числа состояний, нет энтропии — нет и температуры. Для выяснения связи — рассмотрим замкнутую (т. е. не обменивающуюся энергией с окружением) систему. Пусть ее полная энергия равна Е, и пусть эта система находится в равновесии, т. е. все ее микросостояния, имеющие энергию Е, равновероятны (а имеющие другую энергию — имеют нулевую вероятность).
Выделим теперь в этой системе "наблюдаемую малую часть" (например, отдельную молекулу в газе, или макромолекулу с окружающей ее жидкостью). Вся остальная система тогда может рассматриваться как термостат, в который погружена наша "малая часть".
| Замкнутая система, |
Разобьем все микроскопические состояния системы на классы, различающиеся состоянием нашей "малой части". Чем больше микроскопических состояний всей системы в данном классе, — тем больше вероятность его (этого класса) наблюдения, т. е. тем больше вероятность наблюдения данного состояния нашей "малой части".
Пусть наша "малая часть" фиксирована в каком-то микроскопическом состоянии (например, пусть наблюдаемая молекула в газе имеет заданное положение в пространстве и заданную скорость). Пусть она обладает при этом энергией e. Поскольку вся система в целом замкнута, то полная энергия всей системы сохраняется (закон!), и энергия термостата равна E-e. Пусть этой энергии соответствует Mtherm(E-e) возможных микросостояний термостата. Тогда вероятность наблюдения данного состояния нашей "малой части" просто пропорциональна Mtherm(E-e). [Замечание. Мы здесь неявно предположили, что конкретное микросостояние нашей системы не влияет на микросостояния термостата. Строго говоря, это не вполне верно (точнее: это верно только для "термостата", состоящего из разреженного идеального газа), — но учет явлений на границе "нашей системы" и термостата только затемнил бы все дальнейшее изложение. Для совмещения строгости и наглядности — можно представить себе, что наша "наблюдаемая часть" заключена в фиксированный и непроницаемый для молекул термостата объем.]
Если число возможных состояний термостата равно Mtherm(E-e), то логарифм этого числа, по определению, пропорционален энтропии термостата:
Stherm(Е-e) = k ln[Mtherm(E-e)] ; | (8.1) |
коэффициент k здесь вводится только для того, чтобы энтропия, как обычно, измерялась бы в единицах кал/oK; как мы увидим позже, он окажется просто константой Больцмана.
Энергия e "малой части" тоже должна быть относительно мала. Поэтому можно воспользоваться обычным дифференциальным разложением величины Stherm(E-e) по малому параметру e [вы, конечно, помните: f(xo+dx) = f(xo) + (df/dx)|xodx + 1/2(d2f/dx2)|xo(dx)2 + ... = f(xo) + (df/dx)|xodx при малом dx, причем (df/dx)|xo означает, что производная df/dx берется в точке xo]. Итак,
Stherm(E-e) = Stherm(E) — (dStherm/dE)|E. e. | (8.2) |
[Примечание. Так как и S, и Е пропорциональны числу частиц, то dStherm/dE от числа частиц в термостате не зависит, в то время как d2Stherm/dE2 обратно пропорциональна этому числу, т. е. d2Stherm/dE2 ® 0 в очень большом термостате; это и позволяет нам пренебречь членами порядка e2, — а также e3 и т. д. — в формуле 8.2.]
Значит, число возможных микросостояний термостата зависит от энергии e нашей "малой части" как
Mtherm(E-e) = exp[Stherm(E-e)/k] = exp[Stherm(E)/k] xexp[-e. {(dStherm/dE)|E/k}] . | (8.3) |
При этом ни общий множитель exp[Stherm(E)/e] = М(Е), ни число (dStherm/dE)|E не зависит ни от e, ни от конкретного микросостояния нашей "малой части" вообще.
Так как число микросостояний должно расти с энергией (чем больше энергия, — тем большим числом способов ее можно разделить), то формула 8.3 означает просто следующее: чем больше энергии отобрала у термостата наша "малая часть", тем меньше энергии осталось у него, — и тем меньшим числом способов ее можно разделить. При этом падение числа возможных микросостояний термостата (числа способов раздела его энергии) экспоненциально зависит от энергии, содержащейся в нашей "малой части".
Резюмируем. Вероятность наблюдения заданного микросостояния нашей "малой части" (молекулы и т. п.) пропорциональна exp[-e.{(dStherm/dE)|E/k}], где e — энергия этой "малой части", а величина (dStherm/dE)|E/k зависит не от "малой части", а только от окружающей ее среды.
Но, по формуле Больцмана, вероятность пребывания молекулы в заданном состоянии с энергией e пропорциональна exp(-e/kВT) [где T — температура, а kВ — константа Больцмана]! Сравнивая тождественные выражения exp[-e.{(dStherm /dE)|E/k}] и exp(-e/kBT), — видим, что
(dStherm/dE)|E = 1/Т, | (8.4) |
а "k" в формулах 8.1 и 8.3 — это та же константа Больцмана (kB), — если, как обычно, энергия измеряется в джоулях (или в калориях), температура — в оК, а энтропия — в Дж/оК (или в кал/оК).
Формулы 8.1 и 8.4, определяющие температуру как величину, обратную скорости изменения энтропии S (или логарифма числа микросостояний) с энергией Е системы, — основные формулы статистической физики и термодинамики.
Они показывают, в частности, что ln[M(E+kВT)] = S(E+kВT)/k В = [S(E) + (kВТ)(1/T)]/k В = ln[M(E)]+1, — то есть при увеличении энергии на kВT число микросостояний растет в "e" (=2.72) раз, т. е. примерно втрое, — независимо от размеров системы, действующих в ней сил и т. д.
Они означают также, что, зная число микросостояний любой большой системы ("термостата") при разных ее энергиях (или, как говорят, "плотность энергетического спектра" системы), — а точнее, зная зависимость логарифма плотности энергетического спектра системы от энергии, — мы можем найти температуру, соответствующую каждой энергии этой системы. Соответствующий график показан на Рис.8-1.
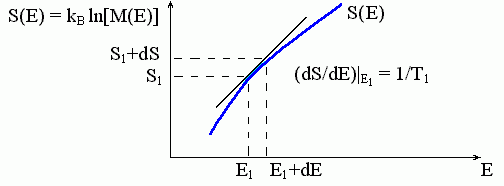
Рис.8-1. Определение температуры большой системы. Зависимость энтропии S этой системы от ее энергии E показана жирной кривой. Наклон этой кривой, dS/dE, определяет температуру T, соответствующую данной энергии E. М(E) — число микросостояний с заданной энергией Е, т. е. плотность спектра энергий состояний системы.
Надо только, чтобы микросостояний было очень много — производную dS/dE можно, естественно, брать только тогда, когда в малом (по сравнению с kBT) интервале энергий находится много микросостояний системы. Именно поэтому температура появляется только в науках, рассматривающих огромное число возможных микросостояний.
Продолжим рассмотрение системы, заключенной в фиксированный и непроницаемый для молекул термостата объем (через стенки которого она, однако, может обмениваться энергией с этим термостатом, температура которого равна Т).
Формулы 8.1 и 8.3 показывают, что вероятность пребывания нашей системы в заданном микросостоянии i с энергией ei при температуре Т есть
wi(Т) = exp(-ei/kBT) / Z(Т), | (8.5) |
где
Z(Т) = Sj exp(-ej/kBT) — | (8.6) |
это нормировочный коэффициент, учитывающий, что сумма вероятностей всех состояний, Sj wj, должна быть равна 1 (сумма Sj здесь и выше берется по всем возможным микросостояниям j рассматриваемой "малой системы").
Величина Z называется статистической суммой для рассматриваемой системы. Зная ее, можно вычислить по формуле 8.5 вероятность каждого из микросостояний этой системы при заданной температуре, — и, далее, ее среднюю энергию при этой температуре
E(Т) = Sjwj. ej | (8.7) |
и ее энтропию
S(Т) = — kBSjwj. ln(wj) . | (8.8) |
Обратите внимание, что формула 8.8 усредняет ln(wj) по всем сосояниям системы, j, с учетом их (состояний) вероятностей wj. Она есть прямое обобщение давно знакомого нам (см. формулу 8.1) определения энтропии S = kB. ln[M(Е)] на случай, когда все состояния системы имеют самые разные вероятности, — а не только две, следовавшие тогда из закона сохранения энергии: wj = 1/M(E) при Ej=Е и wj=0 при Ej ¹ Е.

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 |
