


Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Из сказанного следует, что порядок реакции не может быть предсказан по уравнению реакции, а в каждом отдельном случае должен определяться экспериментально по зависимости скорости реакции от концентрации данного вещества при постоянной концентрации всех остальных веществ.
Порядок реакции является одной из важнейших кинетических характеристик.
12.4. Скорость химической реакции. Время полупревращения
Скорость реакции v - это её интенсивность, выраженная количественно. Она определяется числом молекул, реагирующих в единицу времени в единице объёма. Так как это число эквивалентно изменению количества какого-либо вещества (исходного или продукта), то можно записать:
dn v = ±¾¾¾ , Vdt |
где V - реакционный объём; dn/dt - изменение количества вещества (моль) в единицу времени.
Или, принимая n/V = С,
dС v = ± ¾¾ , (12.1) dt |
где С - концентрация.
Различают истинную (мгновенную) скорость реакции, которая определяется как бесконечно малое изменение концентрации за бесконечно малое время, что математически выражается уравнением (12.1), и среднюю скорость, которая определяется как изменение концентрации за всё время протекания реакции или за какой-либо интересующий экспериментатора отрезок времени:
DС С2 - С1 vср = ± ¾¾ = ± ¾¾¾¾, (12.2) dt t2 - t1 |
где С1 и С2 - концентрации исследуемого вещества, наблюдаемые соответственно начальный и конечный в моменты времени t1 и t2.
Из уравнений (12.1 и 12.2) следует, что если скорость реакции определяется по одному из исходных веществ, разность С2 - С1 будет иметь отрицательный знак, а если по одному из продуктов - то положительный. Скорость реакции всегда положительна, поэтому при вычислении её по исходным веществам в уравнениях (12.1) и (12.2) оставляется знак минус, при вычислении по продуктам - знак плюс.
В общем случае следует различать скорость реакции в целом и скорость реакции по данному веществу. Например, рассмотрим реакцию синтеза аммиака
N2 + 3H2 Û 2NH3 .
Совершенно очевидно, что убыль концентрации водорода в единицу времени втрое превышает убыль концентрации азота, а скорость образования аммиака вдвое больше скорости расходования N2 и в 1,5 раза меньше скорости расходования Н2. Следовательно, скорость реакции по азоту не равна скорости по водороду и скорости по аммиаку.
Размерность скорости химической реакции в соответствии с определением - [концентрация/время]. В системе СИ это моль/м3.·с, в обычной лабораторной практике используют также размерности моль/л·с или моль/л·мин.
В каждом конкретном случае размерность скорости реакции может быть выражена различными единицами в зависимости от самой скорости. Так, например, для реакций разложения лекарственных веществ или коррозии металлов в качестве единицы времени обычно выбирается год, а для реакций, идущих с очень большой скоростью, например, взрывов, реакций нейтрализации и т. п. - доли секунды (милли-, микросекунда и т. д.).
Общая скорость сложной реакции зависит от скорости самой медленной из её стадий (лимитирующая стадия).
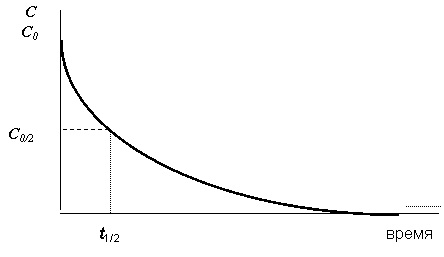
Скорость химических реакций, за исключением реакций нулевого порядка, с течением времени не остаётся постоянной, а замедляется. Причём это замедление носит нелинейный характер, т. е. чем больше времени прошло с начала реакции, тем медленнее она протекает. Средняя скорость за всё время протекания реакции часто бывает очень трудно или даже невозможно определить экспериментально, что связано со сложностью фиксации момента, в который прореагирует последняя молекула одного из исходных веществ. Поэтому в качестве ещё одной кинетической характеристики реакций было введено понятие времени полупревращения t1/2 (в случае реакций разложения называемого также временем полураспада или периодом полураспада). Время полупревращения t1/2 - это время, за которое прореагирует половина взятого для реакции исходного вещества. В отличие от средней скорости за полное время протекания время полупревращения определяется довольно просто и с высокой точностью, что можно проиллюстрировать рис. 12.1.
Рис. 12.1. Зависимость концентрации реагирующих веществ от времени. С0 - начальная концентрация исходного вещества, t1/2 - время полупревращения |
12.5. Закон действующих масс. Константа скорости
В основе учения о химической кинетике лежит её основной постулат, который под названием “закон действующих масс” был открыт бергом и П. Вааге в 1г. г. (В некоторых учебниках можно встретить ошибочное выражение “закон действия масс”, которого следует избегать. и П. Вааге термином “действующая масса” обозначили то понятие, которое сейчас общепринято называют концентрацией. Термин “концентрация” был введён Я. Вант-Гоффом в 90-х годах XIX в.).
В современной формулировке закон читается так:
Скорость химической реакции в каждый момент времени пропорциональна произведению концентраций реагирующих веществ (имеющихся в данный момент времени), возведённых в некоторые степени.
Эти степени не всегда соответствуют стехиометрическим коэффициентам при формулах этих веществ в уравнении реакции, что было проиллюстрировано в п. 12.3.
Например, для произвольной реакции
aA + bB = dD + eE,
когда соотношение концентраций исходных веществ и продуктов близко к соотношению их стехиометрических коэффициентов, математическое выражение закона действующих масс будет выглядеть так:
v = k CaA CbB (12.3)
где CA и CB - концентрации исходных веществ А и В в данный момент времени; а и b - стехиометрические коэффициенты при формулах веществ А и В в уравнении реакции; k - коэффициент пропорциональности, называемый константой скорости данной реакции при данной температуре.
Уточнение о том, что показатели степени при концентрациях реагентов не обязательно совпадают со стехиометрическими коэффициентами, связано с упомянутым выше (п. 12.3) несовпадением порядка и молекулярности реакций. Оно становится понятным, если вспомнить, что показатель степени в уравнении (12.3) представляет собой порядок реакции, который может не соответствовать стехиометрии уравнения реакции и должен для каждого данного случая определяться экспериментально. Поэтому, если соотношения концентраций исходных веществ значительно отличаются от стехиометрии реакции, то степени при концентрациях в уравнении закона действующих масс должны быть соответствующим образом изменены.
Константа скорости k численно равна скорости реакции в стандартном состоянии, то есть при концентрациях всех реагентов, равных 1 в выбранной системе единиц. Она количественно характеризует реакционную способность веществ в элементарной реакции при данной температуре в данной среде.
В случае сложных реакций, в особенности гетерогенных, константа в уравнении закона действующих масс называется коэффициентом скорости или эффективной константой скорости и является эмпирической величиной, физический смысл которой определяется конкретным механизмом реакции.
12.6. Расчёт констант скорости для реакций различных порядков
Наибольшее практическое значение для фармации имеют реакции 1-го и 2-го порядков. Этим порядкам подчиняется подавляющее большинство реакций синтеза и разложения лекарственных веществ, а также реакций, используемых при различных методиках анализа.
12.6.1. Реакции первого порядка
Для произвольной реакции 1-го порядка вида
А ® продукты реакции
уравнение закона действующих масс выглядит так:
dС v = - ¾¾ = kC , dt |
где С - концентрация вещества А. После разделения переменных
dС k dt = - ¾¾ C |
это уравнение можно интегрировать в интервалах = 0 ¸ t, и С0 ¸ Сt:
t Ct dС k ò dt = - ò ¾¾ , 0 C0 C |
получая при этом
k (t - t0) = - (ln Ct - ln C0)
или
k (t - t0) = ln C0 - ln Ct ,
откуда, принимая t0 = 0, получаем окончательное кинетическое уравнение для расчёта константы скорости реакции первого порядка:
1 С0 k = ¾ ln ¾¾ . (12.4) t Ct |
где С0 - начальная концентрация исходного вещества; Ct - концентрация, измеренная в момент времени t.
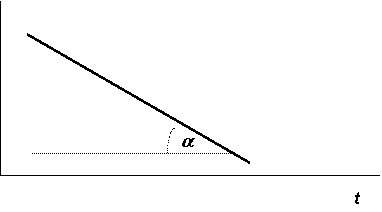
Из этого уравнения следует, что константу скорости реакции 1-го порядка можно определить графически, если построить график зависимости
ln С от времени. При этом тангенс угла наклона графика равен -k (рис. 12.2).
Строя график зависимости ln С от t для неизвестной реакции и получая при этом прямую линию, подобную изображённой на рис. 12.2, можно убедиться в том, что реакция имеет первый порядок.
Константа скорости может быть определена и без построения графика, а именно, как среднее арифметическое из величин, полученных при расчёте по уравнению (12.4) для нескольких текущих концентраций исходного вещества, экспериментально измеренных в различные моменты времени.
Так как в уравнение (12.4) входит отношение начальной и текущей концентраций, то в принципе оно может быть заменено отношением любых
Рис. 12.2. Зависимость ln С - t для реакции первого порядка |
других величин, пропорциональных концентрации (например, углов вращения плоскости поляризации реакционной смеси или её оптической плотности и т. п.), а сама концентрация может быть выражена любыми способами. Часто при вычислениях используют не концентрацию, а количество вещества в молях. Тогда расчётное уравнение для константы скорости будет выглядеть так:
1 а k = ¾ ln ¾¾¾ , (12.5) t а - х |
где а - начальное число молей исходного вещества, x - число молей его, прореагировавшее к моменту времени t.
С помощью уравнений (12.4) и (12.5) могут быть вычислены (если известна константа скорости) и другие важные кинетические характеристики, например, время полупревращения какой-либо реакции или, что представляет особый интерес для фармации, срок годности лекарственных препаратов.
Время полупревращения (t1/2) реакции удобно вычислять с помощью преобразованного уравнения (12.5):
1 а t = ¾ ln ¾¾¾ . k а - х |
Принимая х = а/2, получим:
1 а 1 0,693 t1/2 = ¾ ln ¾¾¾ = ¾ ln 2 = ¾¾¾, (12.6) k а - а/2 k k |
Это же выражение можно получить, используя уравнение (12.4) и принимая Ct = C0/2 .
Из уравнения (12.6) следует, что время полупревращения реакции первого порядка не зависит от начальной концентрации вещества. То есть для уменьшения концентрации, например, от 1 до 0,5 М потребуется ровно столько же времени, сколько для уменьшения концентрации от 0,001 до 0,0005 М. Из уравнения (12.6) следует также, что по экспериментально найденному значению t1/2 можно вычислить константу скорости реакции:
0,693 k = ¾¾¾ . t1/2 |
Реакции разложения многих лекарственных веществ, особенно реакции, идущие в растворах, являются реакциями первого или псевдопервого порядка, поэтому срок годности их тоже может быть рассчитан по кинетическому уравнению (12.5). Согласно действующей нормативной документации лекарственный препарат считается годным к употреблению, если содержание лекарственного вещества в нём отвечает требованиям соответствующей фармакопейной статьи. Принимая, что в свежеприготовленном препарате количество наиболее быстро разлагающегося вещества а = 100%, а допустимый процент разложения лекарственного вещества равен х, можно, зная константу скорости разложения данного вещества при температуре его хранения, по уравнению
1 100 t = ¾ ln ¾¾¾¾ . k 100 - х |
вычислить ориентировочный срок его годности.
12.6.2. Реакции второго порядка
Это самый распространенный тип реакций, которые могут протекать по схемам
1) А + В ® продукты реакции
или 2) 2А ® продукты реакции
Скорость таких реакций может быть описана такими уравнениями закона действующих масс:
для 1-го типа -
dС v = - ¾¾ = kCACB, dt |
а для 2-го -
dС v = - ¾¾ = kC2 . dt |
Далее мы будем рассматривать, главным образом, реакции, относящиеся к 1-му типу, как наиболее часто встречающиеся. Хотя следует помнить о том, что и реакции 2-го типа довольно широко распространены. В частности, разложение некоторых лекарственных веществ может протекать именно по этому типу.
Время, за которое вступает во взаимодействие определённая доля начального количества веществ, в реакциях второго порядка зависит от соотношения начальных концентраций исходных веществ. Поэтому принято рассматривать два возможных случая: 1) C0 A = C0 B и 2) C0 A ¹ C0 B.
При равных начальных концентрациях исходных веществ можно принять, что C0 A = C0 B = C0. При этом в каждый момент времени будет соблюдаться равенство Ct A = Ct B = Ct . Кинетическое уравнение для данного случая выглядит так:
dС v = - ¾¾ = kC2. dt |
Разделяя переменные
dС - ¾¾ = k dt C2 |
и интегрируя в интервалах = 0 ¸ t, и С0 ¸ Сt:
t Ct dС k ò dt = - ò ¾¾ , 0 C0 C2 |
получаем
1 1 Ct - C0 kt = ¾ - ¾ = ¾¾¾¾ C0 Ct C0 Ct |
и окончательно
1 Ct - C0 k = ¾ · ¾¾¾¾ (12.7) t C0 Ct |
Заменяя концентрации на соответствующие количества вещества в моль
(C0 = а, Ct = а - х, и Ct - C0 = х), получим
1 х k = ¾ · ¾¾¾¾ . (12.8) t а(а - х) |
При различных начальных концентрациях исходных веществ, когда C0 A ¹ C0 B и Ct A ¹ Ct B, интегрирование кинетического уравнения
dС v = - ¾¾ = k CA CВ dt |
или
dС k dt = - ¾¾¾ CA CÂ |
даёт выражение
1 C0 B Ct A k = ¾¾¾¾¾¾¾ ln ¾¾¾¾ . (12.9) t (C0 A - C0 B) C0 A Ct В |
Заменяя концентрации на количество вещества (моль), получим
1 b(a - x) k = ¾¾¾¾ ln ¾¾¾¾ . (12.10) t (а - b) a(b - x) |
где а и b - соответственно начальное число молей веществ А и В; x - количество вещества, прореагировавшее к моменту времени t.
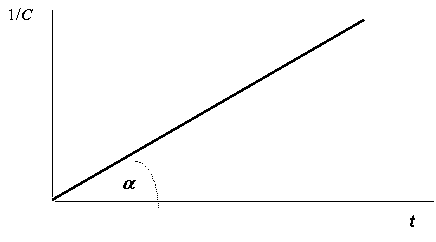
График зависимости 1/С от t для реакций второго порядка представляет собой прямую, проходящую через начало координат, с тангенсом, равным k; это означает, что константа скорости реакции второго порядка тоже может быть определена графически (рис. 12.3):
Рис. 12.3. Зависимость 1/С - t для реакции второго порядка |
Время полупревращения для реакции второго порядка рассчитывается с помощью выведенных уравнений по известной константе скорости. Для случая одинаковых начальных концентраций исходных веществ
1 1 t1/2 = ¾¾ или t1/2 = ¾¾ , k C0 k а |
для случая различных начальных концентраций
1 1 t1/2 = ¾¾¾¾¾¾ или t1/2 = ¾¾¾¾ . k C0 А - C0 В k (а - b) |
То есть, в отличие от реакций первого порядка, в данном случае время полупревращения зависит от начальной концентрации исходных веществ.
12.7. Определение порядка реакции
В соответствии с вышеизложенным порядок простой необратимой реакции может быть определён такими способами:
1) Интегральные методы.
1.1) По константам скорости. Процедура состоит в измерении концентрации реагента (или реагентов) в различные моменты времени и в подстановке полученных данных в уравнения для расчёта константы скорости реакции первого и второго порядка, выведенные ранее (а при необходимости и в уравнение для третьего порядка). То уравнение, которое для различных интервалов времени даёт значения константы скорости, характеризующиеся наименьшим разбросом значений, следует считать наиболее правильно отражающим порядок реакции. Если вычисления ни по одному из этих уравнений не дают удовлетворительных результатов (значения константы скорости во всех случаях имеют значительный разброс), то считается, что реакция имеет дробный порядок. В том случае, когда концентрация исходных веществ линейно уменьшатся с течением времени, принимается, что реакция имеет нулевой порядок.
1.2) По времени полупревращения. Метод аналогичен предыдущему, с тем различием, что в этом случае рассчитываются не k, а величины t1/2.
1.3) Графический. По экспериментальным данным строятся графики зависимостей для первого порядка ln C - t (см. рис. 12.2), для второго порядка 1/C - t (см. рис. 12.3), при необходимости для нулевого порядка C - t и для третьего порядка 1/C2 - t. Тот график, на котором точки лучше всего укладываются на прямую линию, соответствует порядку реакции.
2) Дифференциальный метод (по Вант-Гоффу, 1884)
Уравнение закона действующих масс для исследуемой реакции можно приблизительно записать в виде
v = kCn,
где n - суммарный порядок реакции. Логарифмируя это выражение, получим уравнение прямой, не проходящей через начало координат:
lg v = lg k + nlg C.
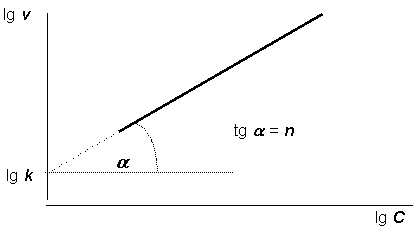
Построив по экспериментальным данным график зависимости логарифма скорости исследуемой реакции от логарифма концентрации одного из исходных веществ (рис. 12.4), можно определить её порядок как тангенс угла наклона графика к оси абсцисс. При этом от оси ординат отсекается отрезок, равный логарифму константы скорости реакции.
Рис. 12.4. Определение порядка реакции методом Вант-Гоффа |
12.8. Механизмы химических реакций
Большинство химических реакций являются сложными, то есть они протекают в несколько стадий. Одной из задач кинетики является определение числа и вида промежуточных стадий, потому что только так можно представить, как протекает реакция в целом. Отдельные стадии называются элементарными реакциями. Совокупность элементарных реакций представляет собой механизм суммарной реакции. При рассмотрении механизма говорят о молекулярности каждой из её стадий, которая определяется числом молекул или других частиц – атомов, ионов, радикалов, - участвующих в данной элементарной реакции. Отдельные стадии являются моно-, би - или тримолекулярными в зависимости от того, одна, две или три частицы вступают во взаимодействие на данной стадии. Для элементарных реакций молекулярность совпадает с их порядком, но по отношению к суммарной реакции эти термины не являются синонимами.
Процесс установления механизма какой-либо реакции нельзя описать кратко, поскольку он связан со многими экспериментальными трудностями. Кинетические данные об изменении концентрации во времени оказываются очень полезными при определении механизма. Некоторые сведения о механизме можно также получить, используя радиоактивные изотопы для определения последовательных состояний различных атомов или молекул в ходе реакции и спектроскопические методы для идентификации промежуточных соединений.
Однако подробное рассмотрение сложных реакций и способов установления их механизма не входит в программу нашего курса.
12.9. Влияние температуры на скорость реакции
Скорость реакции зависит от многих факторов, в том числе от природы реагирующих веществ, природы среды, концентрации (влияние которой рассмотрено ранее) и др. В ряде случаев, в особенности для цепных реакций, скорость может определяться даже размерами сосуда, в котором она происходит. Однако с теоретической и с практической точек зрения наряду с концентрацией особенно важным является влияние на скорость реакции температуры.
Для большинства химических реакций (за исключением тримолекулярных и ферментативных) скорость с ростом температуры увеличивается. В принципе это влияние температуры может быть сопряжено с изменением трёх величин: концентрации реагирующих веществ, константы скорости и порядка реакции. Но концентрация веществ (особенно для реакций в жидкой или твёрдой фазе) с температурой изменяется мало, а если и изменяется, то, как правило, в сторону уменьшения; изменение же порядка реакции под влиянием температуры - довольно редкое явление. Вследствие этого в химической кинетике изменение скорости реакции под влиянием температуры в первую очередь связывается с изменением константы скорости реакции.
12.9.1. Правило Вант-Гоффа
Наглядное представление о зависимости скорости химических реакций от температуры даёт правило Вант-Гоффа, согласно которому
при повышении температуры на 10 градусов скорость реакции увеличивается в 2 - 4 раза.
Это правило первоначально было установлено для реакций, протекающих в растворах при невысоких температурах, а затем было распространено и на другие реакции. Оно связано с понятием температурного коэффициента скорости реакции g
kT+10 g = ¾¾¾ , kT |
где kT и kT+10 - константы скорости исследуемой реакции при какой-то данной температуре Т и при температуре, превышающей Т на 10 градусов.
При температурах, близких к комнатной, для большинства газовых реакций и реакций в растворах g » 3. Использование величины g позволяет приближённо оценить изменение скорости реакции при некотором увеличении температуры на 10n градусов, причем число n может быть как целым, так и дробным:
v2 kT + 10n ¾ = ¾¾¾ = gn, v1 kT |
где v1 и v2 - скорости одной и той же реакции при температурах соответственно Т и Т + 10.
При очень высоких и очень низких температурах правило Вант-Гоффа не выполняется.
12.9.2. Теория активных столкновений. Уравнение Аррениуса
Количественная зависимость константы скорости реакции от температуры была впервые предложена тоже Я. Вант-Гоффом (1887) в виде уравнений изохоры и изобары химической реакции (см. п. 4.5.2).
Эта идея была развита С. Аррениусом (1889), который открыл, что температурную зависимость скорости многих реакций можно описать уравнением:
k = Ае-Е*/RT
где k - константа скорости, e - основание натуральных логарифмов, R - универсальная газовая постоянная, T – температура, А - предэкспоненциальный множитель, Е* - энергия активации реакции.
Чтобы выяснить физический смысл величин А и Е*, входящих в уравнение Аррениуса, следует сначала познакомиться с основными положениями теории активных соударений (столкновений) (С. Аррениус и Я. Вант-Гофф; 1880-е г. г.):
1) Химическое взаимодействие между молекулами возможно только при их столкновении.
2) Не каждое столкновение молекул приводит к химическому взаимодействию, т. е. является результативным или, по терминологии Аррениуса, активным. Существует некий энергетический барьер, преодолеть который и вступить во взаимодействие может лишь часть молекул, причём, как правило, это очень малая часть от их общего числа в системе.
3) Причиной, обусловливающей существование энергетического барьера, является взаимное отталкивание электронных оболочек молекул при их сближении.
Когда две частицы удалены друг от друга на очень большое расстояние, между ними нет никакого взаимодействия и потенциальная энергия такой системы равна нулю. При меньших расстояниях между частицами они притягиваются друг к другу, и потенциальная энергия системы понижается. При дальнейшем уменьшении расстояния становятся заметными силы отталкивания электронных оболочек молекул и потенциальная энергия резко возрастает. Поэтому для сближения частиц до расстояния, на котором начнется перераспределение электронов на их орбиталях (т. е. химическое взаимодействие), частицы должны обладать достаточно большим запасом кинетической энергии. Силы отталкивания между частицами и представляют между собой так называемый потенциальный или энергетический барьер, а химическое взаимодействие возможно только в том случае, если сталкивающиеся молекулы способны преодолеть его.
4) Для того, чтобы молекулы могли при столкновении преодолеть энергетический барьер, они должны двигаться навстречу друг другу с достаточно большой скоростью. Для достижения этой необходимой скорости нужна определённая энергия, называемая энергией активации. Энергия активации Е* - это избыток энергии активных молекул по сравнению с неактивными, или иначе, энергия, которой должны обладать молекулы, чтобы иметь возможность вступить во взаимодействие. Размерность СИ энергии активации - Дж/моль.
5) Чем больше энергия активации реакции, тем больше энергетический барьер, и тем меньшее число молекул способно его преодолеть. Поэтому, чем больше Е*, тем медленнее идёт реакция.
6) С повышением температуры увеличивается скорость теплового движения молекул, поэтому доля активных молекул возрастает. Иными словами, при повышении температуры происходит термическая активация, приводящая к увеличению скорости реакции.

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 |
