


Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
откуда, принимая, что суммарное число молей всех компонентов равно 1, получаем для работы расширения
pdV = (RT/V)dV ,
Так как максимальная полезная работа может быть вычислена при интегрировании выражения: V2
w’max = ò pdV ,
V1
получаем
V2 RT V2 р2 w’max = ò ¾¾ dV = - RT ln ¾¾ = RT ln ¾¾ = RT(ln р2 - ln р1), V1 V V1 р1 |
где p1 - давление исходной газовой смеси, р2 - равновесное давление.
Заменяя ln р1 суммой логарифмов начальных парциальных давлений, а ln p2 - суммой логарифмов равновесных парциальных давлений веществ, получим:
w’max = RT [(ln pDdравн + ln pEeравн - ln pAaравн - ln pBbравн) -
- (ln pDdнач + ln pEeнач - ln pAaнач - ln pBbнач)],
откуда
рDd рЕe рDd рЕe w’max = RT [ln(¾¾¾¾)равн - ln(¾¾¾¾)нач], рAa рBb рAa рBb |
или
рDd рЕe w’max = RT [ln Кр - ln(¾¾¾¾)нач], (4.1) рAa рBb |
а так как w’max = -DGr ,
то можно записать:
рDd рЕe DGr = -RT [ln Кр - ln(¾¾¾¾)нач], (4.2) рAa рBb |
Для процессов, идущих при постоянном объёме, можно получить аналогичные выражения, в которые входит максимальная работа и изменение энергии Гельмгольца в ходе реакции. При этом парциальные давления заменяются начальными концентрациями веществ:
СDd СЕe wmax = RT [ln Кс - ln(¾¾¾¾)нач], (4.3) СAa СBb |
CDd CЕe DAr = -RT [ln Кc - ln(¾¾¾¾)нач], (4.4) CAa CBb |
Уравнения (4, выведенные Я. Вант-Гоффом, называются уравнениями изотермы химической реакции. Они дают возможность определить, в каком направлении и до какого предела может протекать реакция в рассматриваемых условиях при заданном составе реакционной смеси при постоянной температуре.
Для стандартных условий, когда исходные парциальные давления (или исходные концентрации или активности) всех веществ-участников реакции равны единице, уравнения изотермы будут выглядеть так:
w’max = RT ln Kp ; DGor = - RT ln Kp (4.5)
и
wmax = RT ln Kс ; DАor = - RT ln Kс .
Отсюда следует, что определяя стандартную величину DGor или DАor для реакции, можно легко вычислить её константу равновесия. (Способ расчёта DGor и DАor был показан в п. 3.8).
4.5.2. Влияние температуры на равновесие. Уравнения изобары
и изохоры химической реакции
d(RT ln Kp) - RT ln Kp - DH - ¾¾¾¾¾¾ = ¾¾¾¾¾¾¾¾ ; dT T |
Для вывода уравнения изобары подставим соотношение (4.5) в уравнение Гиббса - Гельмгольца (3.11)
Заменим знаки “минус” в обеих частях уравнения на “плюс”:
d(RT ln Kp) RT ln Kp + DHr ¾¾¾¾¾¾ = ¾¾¾¾¾¾¾ ; dT T |
после чего дифференцируем левую часть по температуре:
RT dln Kp RT ln Kp + DHr 0 + RT ln Kp + ¾¾¾¾¾ = ¾¾¾¾¾¾¾¾ ; dT T |
Умножая обе части уравнения на Т и сокращая, получим:
RT2 d ln Kp ¾¾¾¾¾¾ = DHr dT |
или
d ln Kp DHr ¾¾¾¾ = ¾¾ (4.6) dT RT2 |
Для реакций, идущих в изохорных условиях можно получить аналогичное выражение
d ln Kc DUr ¾¾¾¾ = ¾¾ (4.7) dT RT2 |
Уравнения (4.6) и (4.7) тоже были выведены Я. Вант-Гоффом и названы соответственно уравнениями изобары и изохоры химической реакции. Они показывают зависимость константы равновесия химической реакции от температуры.
Уравнения изотермы, изобары и изохоры химической реакции в количественной форме отображают принцип подвижного химического равновесия Ле-Шателье - Брауна. С их помощью можно рассчитать условия, при которых константа равновесия будет соответствовать увеличению выхода требуемого продукта, например, лекарственного вещества. Если процесс экзотермический (DH < 0), увеличение температуры будет приводить к уменьшению отношения DH/RT2, а значит, и к уменьшению константы равновесия, т. е. в конечном итоге к уменьшению выхода продуктов. В случае эндотермического процесса наблюдается обратная зависимость.
4.5.3. Влияние на равновесный выход изменения объёма
и давления реакционной смеси
Для реакций, идущих в газовой фазе, об изменении объёма реакционной смеси можно судить по изменению числа молей реагирующих веществ
Dn = åni прод - åni исх
Возможны три случая, соответствующих различным типам химических реакций:
а) Dn < 0 (реакция идет с уменьшением объёма). Например, реакция синтеза аммиака:
N2 (г) + 3H2 (г) Û 2NH3 (г) ; Dn = + 3) = -2
В соответствии с принципом Ле-Шателье уменьшение объёма (при увеличении давления) будет сдвигать равновесие этой и подобных реакций вправо, а увеличение объёма (при уменьшении давления) - влево.
б) Dn > 0 (реакция идет с увеличением объёма). Например, реакция разложения метанола:
CH3OH (г) Û CO (г) + 2H2 (г) ; Dn = (1 += 2
В этом случае уменьшение объёма (или увеличение давления) будет сдвигать равновесие влево, а увеличение объёма (при уменьшении давления) - вправо.
в) Dn = 0 (реакция идет без изменения объёма). Например, реакция хлора с бромоводородом:
Cl2 (г) + 2HBr (г) Û Br2 (г) + 2HCl (г) ; Dn = (1 ++ 2) = 0
На выходе продуктов таких реакций изменение объёма (давления) реакционной смеси не сказывается.
4.6. Способы вычисления констант равновесия
а) Интегрируя после разделения переменных уравнение изобары (4.6) в предположении, что тепловой эффект реакции мало зависит от температуры
2 DHr Т2 dT ò d ln Kp = ¾¾ ò ¾¾ 1 R Т1 T2 |
получаем
Kp 2 DHr 1 1 ln Kp 2 - ln Kp 1 = ln ¾¾ = ¾¾ (¾ - ¾) Kp 1 R Т1 T2 |
или
Kp 2 DHr Т2 - Т1 ln ¾¾ = ¾¾ (¾¾¾¾) , Kp 1 R Т1 T2 |
где Kp 1 и Kp 2 - константы равновесия реакции при температурах Т1 и T2 соответственно; DHr - изобарный тепловой эффект реакции в интервале температуры Т1 ¸ T2.
С помощью этого уравнения по DHr и одной из Кр (как правило, по стандартному тепловому эффекту DHоr 298, вычисленному по справочным данным, и Кр 1 при температуре Т1 = 298 К) можно вычислить другую константу равновесия Кр 2 при любой другой температуре Т2. С другой стороны, с помощью двух констант равновесия Кр 1 и Кр 2 при двух температурах Т1 и Т2 можно рассчитать средний тепловой эффект реакции. Необходимо помнить, что этот способ вычисления Кр применим в не очень большом интервале температуры, когда зависимостью теплового эффекта от температуры можно пренебречь.
Для изохорных условий, например, для реакций, идущих в растворах, можно получить аналогичное выражение, в которое входят константы равновесия Кс и изохорный тепловой эффект DUоr :
Kc 2 DUr Т2 - Т1 ln ¾¾ = ¾¾ (¾¾¾¾) , Kc 1 R Т1 T2 |
б) Как было показано ранее, при стандартных условиях имеет место равенство
DGor = - RT ln Kp .
В то же время изменение энергии Гиббса для реакции связано с её тепловым эффектом при постоянном давлении и изменением энтропии уравнением
DGor = DHor - TDSor .
Объединяя два этих выражения, получаем
-RT ln Kp = DHor - TDSor ,
откуда следует, что
DSor DHоr ln Kp = ¾¾ - ¾¾ . R RТ |
Это выражение называется уравнением Вант-Гоффа. С его помощью по стандартным значениям DHor и DSor, (например, вычисленным по справочным значениям), можно рассчитать константу равновесия реакции для
Т = 298 К или для другой температуры, не очень намного отличающейся от 298 К.
С другой стороны, если известны значения Kp в некотором небольшом интервале температур, при посредстве уравнения Вант-Гоффа можно графическим способом определить величины DHor и DSor. График зависимости логарифма константы равновесия от 1/T (рис. 4.1) представляет собой прямую линию с отрицательным углом наклона, причем тангенс угла наклона её к оси абсцисс равен -DHor/R, а точка пересечения графика с осью ординат дает DSor/R:
|
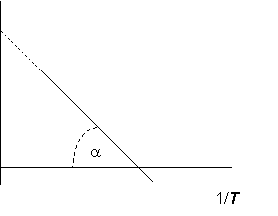
Рис. 4.1. Зависимость ln Kp - 1/T для обратимой реакции
Следует помнить, что описанные способы вычисления константы равновесия химической реакции применимы лишь в достаточно узком интервале температуры. Когда разность Т2 - Т1 превышает несколько десятков градусов, требуется учитывать зависимость теплового эффекта реакции и теплоёмкости системы от температуры, что приводит к более сложным, но дающим более точные результаты уравнениям.
4.7. Химическое равновесие в гетерогенных системах
Рассмотренные ранее закономерности относятся, главным образом, к гомогенным реакциям, т. е. к реакциям с участием веществ, находящихся в одном физическом состоянии - в виде газа или в виде раствора. Равновесия, в которых принимают участие вещества, находящиеся в двух или нескольких физических состояниях (например, газ с жидкостью или с твёрдым веществом), называются гетерогенными равновесиями.
В качестве примера рассмотрим разложение карбоната кальция CaCO3, используемого в фармации в качестве антацидного средства. Это удобная модель для рассмотрения разложения различных твёрдых веществ, в том числе и лекарственных, идущего с образованием газообразных продуктов:
CaCO3 (т) Û CaO (т) + CO2 (г)
В соответствии с законом действующих масс выражение для константы равновесия этой реакции можно написать так:
pCaO pCO2 Kр = ¾¾¾¾¾ pCaCO3 |
Парциальные давления CaO и CaCO3 в газовой фазе, во-первых, очень малы, а во-вторых, остаются практически постоянными в любой момент протекания реакции. Это значит, что пока твёрдые CaCO3 и CaO находятся в контакте с газом, их влияние на равновесие будет неизменным. В этом случае константа равновесия не зависит от количества твёрдой фазы. Можно разделить обе части выражения для константы равновесия на величину pCaO/pCaCO3 и принять, что
K’p = pCO2 ,
где K’p = KppCaC03/pCaO - модифицированная константа равновесия; при этом парциальные давления CaCO3 и CaO входят в величину K’p в неявном виде.
Если парциальное давление СО2 над CaCO3, при данной температуре поддерживается меньшим, чем значение K’p, то весь CaCO3 превратится в CaO и CO2; если же парциальное давление pCO2 больше, чем K’p, то весь СaO превратится в CaCO3. Равновесное же парциальное давление CO2, равное K’p при данной температуре, называется давлением диссоциации.
При достижении давления СО2 1 атм равновесие в данной реакции сдвигается в сторону диссоциации СаСО3, т. е. разложения карбоната кальция. Приведённая ниже таблица показывает, что это происходит при температуре 897оС:
Давление CO2 над CaCO3
Температура, оС Давление, атм
500 9,30´10-5
600 2,42´10-3
700 2,92´10-2
800 0,220
897 1,000
1000 3,871
1100 11,50
1200 28,68
Подобные рассуждения и понятие давления диссоциации могут быть распространены и на другие гетерогенные реакции с участием твёрдых веществ. В том случае, когда лекарственное вещество (в порошке или в таблетках) может реагировать с газами, находящимися в воздухе (H2O, O2, CO2), или разлагаться с их выделением, необходимо следить, чтобы парциальное давление этих газов и паров в атмосфере склада было меньше, чем давление диссоциации (или соответствующая константа равновесия K’p).
III. Ф А З О В О Е Р А В Н О В Е С И Е
ГЛАВА 5
ТЕРМОДИНАМИКА ФАЗОВОГО РАВНОВЕСИЯ
5.1. Краткий исторический очерк
Один из главных физических законов, лежащий в основе кинетической теории газов, - закон Р. Бойля (1662) - Э. Мариотта (1676) был сформулирован во 2-й половине
XVII в.
В России первые исследования, относящиеся к фазовому равновесию, были выполнены , который, изучая свойства растворов, установил, что повышение концентрации растворённого вещества вызывает понижение температуры его замерзания.
В начале XVIII в. идут изыскания в области термометрии - создаются различные конструкции термометров и предлагается большое число температурных шкал, из которых наибольшее признание получили шкалы (1714), (1730) и, в особенности А. Цельсия (1744). В конце XVIII - начале XIX в. открываются другие главные газовые законы – Ж. А.С. Шарля (1787), Дж. Дальтона (1, -Люссака (1, А. Авогадро (1811).
В 1834 г. выходит одна из основополагающих для физики и химии работа Б. П.Э. Клапейрона, в которой были даны обобщённое уравнение состояния идеального газа, впоследствии усовершенствованное делеевым, а также основное уравнение фазового равновесия однокомпонентных систем. В 1848 У. Томсон (Кельвин) вводит абсолютную шкалу температуры.
Большой вклад в учение о фазовом равновесии и, в особенности, в теорию растворов в 1х гг. внёс . Он ввёл понятие критической температуры жидкостей, исследовал явления изоморфизма твёрдых тел, сконструировал пикнометр. Он же создал гидратную теорию растворов, нашёл общее уравнение состояния идеального газа (уравнение Менделеева - Клапейрона).
В работах Дж. У.Гиббса 1гг. открыт фундаментальный закон фазового равновесия - правило фаз. В 1880-е гг. открыты основные законы равновесия в системах “жидкость - пар” (законы и ). Тогда же были проведены исследования по связи температуры замерзания и закипания растворов с концентрацией, а Я. Вант-Гоффом открыты закономерности осмотического давления; Д. П.Коноваловым созданы теоретические основы перегонки жидких смесей. Курнакова создано новое направление - физико-химический анализ, позволивший проводить систематическое изучение сложных многокомпонентных систем.
Современные исследования в области фазового равновесия в основном связаны с анализом свойств многокомпонентных систем и с явлением полиморфизма твёрдых веществ.
5.2. Фазовые переходы
Законы и закономерности, изучаемые в данном разделе, являются теоретической основой таких процессов, как экстракция, широко применяемая в фармации для извлечения биологически активных и лекарственных веществ из природного сырья, из растворов при промышленном получении, приготовление растворов и осаждение веществ из них, перегонка, сушка твёрдых веществ и растворителей, физическое взаимодействие веществ в лекарственных формах. Кроме того, учение о фазовых равновесиях необходимо для выяснения причин физической совместимости или несовместимости лекарственных веществ.
Фазовые переходы - это переходы вещества из одного фазового состояния в другое при изменении параметров, характеризующих термодинамическое равновесие. Такие широко распространённые процессы, как плавление, кипение, испарение, возгонка (сублимация), растворение, и обратные им (отвердевание, конденсация паров, кристаллизация из растворов), являются фазовыми переходами. К фазовым переходам относятся также полиморфные превращения, т. е. переходы сложных веществ из одного кристаллического состояния в другое, аллотропные переходы твёрдых простых веществ, жидкостная экстракция и ряд других.
5.3. Основные понятия
Фаза Ф - совокупность частей системы, тождественных по химическому составу и физическим свойствам, находящихся между собой в термодинамическом равновесии и отделённых поверхностями раздела от других частей. Всякая гомогенная система однофазна, т. е. характеризуется отсутствием внутренних поверхностей раздела. Гетерогенная система содержит несколько фаз (как минимум, две). В гетерогенной системе фазы имеются внутренние поверхности раздела (иногда называемые межфазными границами).
Фаза - более общее понятие, чем индивидуальное вещество или агрегатное состояние. Во многих случаях одно и то же вещество может находиться в системе в виде различных фаз. Так, жидкая вода в равновесии со льдом и паром образует трёхфазную систему; в системе, состоящей из кристаллов CuSO4·5H2O, погружённых в насыщенный раствор CuSO4, вода входит в состав и жидкой фазы (раствора), и твёрдой (кристаллов). Простые фазы содержат одно химическое вещество, сложные, например, растворы, состоят из нескольких веществ.
Фазовое равновесие - сосуществование термодинамически равновесных фаз, образующих гетерогенную систему.
Компонент - индивидуальное химическое вещество, входящее в состав системы. Компонентом считается только то вещество, которое в принципе может быть выделено из системы и может существовать самостоятельно в течение достаточно длительного времени. Например, водный раствор углекислого газа содержит кроме Н2О и СО2 ещё и угольную кислоту Н2СО3. Однако угольная кислота существует только в растворе и не может быть из него выделена. Значит, компонентами данной системы являются только Н2О и СО2. Не являются компонентами и ионы, образующиеся при диссоциации электролитов в растворах.
Число независимых компонентов системы К – это число компонентов, необходимых для создания полного состава системы. Оно равно общему числу компонентов минус число химических реакций, протекающих между ними. Например, для системы, состоящей из карбоната кальция, углекислого газа и оксида кальция, число независимых компонентов равно 2, так как при помещении в замкнутый сосуд любых двух из этих веществ третье появится самопроизвольно из-за протекающей обратимой реакции.
5.4. Правило фаз
Наиболее общий закон, описывающий фазовые равновесия, - это правило фаз, являющееся следствием второго начала термодинамики. Правило фаз, открытое Дж. У.Гиббсом в 1876 г., связывает число фаз, находящихся в равновесии, число независимых компонентов и число параметров, необходимых для полного описания системы:
Число степеней свободы (вариантность) термодинамической системы, находящейся в равновесии, на которую из внешних факторов влияют только давление и температура, равно числу независимых компонентов минус число фаз плюс два:
С = К - Ф + 2
Если состояние системы зависит не от двух внешних факторов, то в общем виде выражение для правила фаз выглядит следующим образом:
С = К - Ф + n,
где n – число факторов, определяющих состояние. Например, на системы, состоящие только из конденсированных (твёрдых или жидких) фаз, давление практически не оказывает влияния, и поэтому для них n = 1.
Вариантность системы С может быть представлена как число внешних условий (температура, давление, концентрация и др.), которые экспериментатор может изменять, не изменяя при этом числа фаз в системе. Необходимость таких воздействий на систему связана с интенсификацией желательных процессов (например, ускорение экстракции, растворения и т. п.) или, наоборот, с замедлением нежелательных.
Правило фаз показывает, что число степеней свободы возрастает с увеличением числа компонентов и с уменьшением числа фаз системы. В зависимости от состава системы при расчёте по правилу фаз можно получить различные значения вариантности.
При С = 0 система называется нонвариантной (или инвариантной); изменение любого параметра состояния приводит к изменению числа фаз.
При С = 1 система называется моновариантной; лишь какой-либо один из параметров может быть изменён без изменения числа фаз, находящихся в равновесии.
При С = 2 система называется бивариантной, при С = 3 - тривариантной. В таких системах без изменения числа фаз, находящихся в равновесии, можно изменять в определённых интервалах соответственно два или три параметра.
5.5. Общее условие фазового равновесия. Химический потенциал
Система будет находиться в состоянии фазового равновесия (т. е. ни один из её компонентов не будет переходить из одной фазы в другую) при условии, что химический потенциал каждого компонента во всех фазах является одинаковым.
d (DG i) mi = (¾¾¾¾)p, T dni |
Химический потенциал m i-того компонента - это производная энергии Гиббса данного компонента по его количеству вещества:
Иными словами, химический потенциал i-того компонента равен приращению энергии Гиббса при добавлении одного моля этого компонента к большому объёму системы (настолько большому, что состав системы при этом практически не изменяется) при постоянных температуре и давлении. Химический потенциал - важная термодинамическая функция. С ним связаны такие понятия, как работа расширения идеального и реального газов, активность и коэффициент активности компонентов растворов. На основе химического потенциала того или иного компонента выводятся условия его существования в составе определённой фазы или возможность перехода из одной фазы в другую.
Характерным свойством химического потенциала является то, что он равен производной по количеству вещества всех термодинамических функций:
d (DGi) d (DHi) d (DUi) d (DA) m i = (¾¾¾¾)p, T = (¾¾¾¾)p, T = (¾¾¾¾)V, T = (¾¾¾¾)V, T dni dni dni dni |
ГЛАВА 6
ФАЗОВЫЕ РАВНОВЕСИЯ В ОДНОКОМПОНЕНТНЫХ
СИСТЕМАХ
6.1. Связь между давлением и температурой фазовых переходов.
Уравнение Клапейрона
В однокомпонентных системах, т. е. системах, состоящих из одного вещества, возможны такие фазовые переходы: плавление твёрдого тела и отвердевание жидкости (кристаллизация, “замерзание”), испарение жидкости и конденсация пара в жидкость, возгонка (сублимация) и конденсация пара в твёрдое тело, а также аллотропные и полиморфные переходы (переходы твёрдых веществ из одной кристаллической модификации в другую).
Когда две фазы (a и b) чистого вещества в однокомпонентной системе находятся в равновесии, их химические потенциалы при данных температуре и давлении одинаковы:
ma = mb
Если при постоянном давлении изменять температуру или при постоянной температуре изменять давление, то компонент будет переходить из одной фазы в другую. Например, при плавлении льда вода переходит из твёрдой фазы в жидкую, а при кипении воды - из жидкой фазы в паровую. В пределе это может привести к исчезновению одной из фаз. Но если одновременно изменять и температуру, и давление таким образом, чтобы химические потенциалы двух фаз оставались одинаковыми, то в системе по-прежнему будут в равновесии находиться обе фазы.
Уравнение, показывающее связь между изменениями температуры и давления в однокомпонентной системе при сохранении неизменного числа фаз, вывел Б. П.Э. Клапейрон (1834).
При изменении Т и р должно соблюдаться равенство
dma = dmb
и, значит,
dGa = dGb
Поскольку G, как и m, зависит только от давления и температуры, можно это равенство записать в виде:
dGa dGa dGb dGb (¾¾¾)T dp + (¾¾¾)p dT = (¾¾¾)T dp + (¾¾¾)p dT . (6.1) dp dT dp dT |
Из фундаментального уравнения
dU = TdS - pdV ,
полученного из первого начала термодинамики, можно получить соотношения
dG dG (¾¾¾)T = V и (¾¾¾)p = - S dp dT |
Подставляя их в ранее полученное выражение (6.1), получаем:
Vadp - SadT = Vbdp - SbdT
или
dp Sb - Sa DS ¾¾ = ¾¾¾¾ = ¾¾ . dT Vb - Va DT |
Так как при равновесии
DG = DH - TDS = 0,
то DS = DH /T
и, значит,
dp DН ¾¾ = ¾¾¾ . dT TDV |
Это уравнение - уравнение Клапейрона - применимо ко всем фазовым переходам - процессам испарения, возгонки, плавления и обратным им, а также к полиморфным превращениям чистого вещества. Производная dp/dT показывает, как изменяется давление, при котором происходит фазовый переход, при изменении температуры.

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 |
