


Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Возвращаясь к уравнению Аррениуса, отметим, что величина e-Е*/RT (“экспонента”) равна доле активных молекул, обладающих избыточной энергией Е* для вступления в химическое взаимодействие, а коэффициент А (предэкспоненциальный множитель) равен полной частоте соударений между молекулами реагирующих веществ в реакционном объёме.
Логарифмируя уравнение Аррениуса, получим уравнение прямой, не проходящей через начало координат:
E* ln k = ln A - ¾¾ . RT |
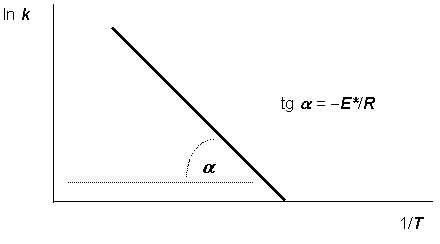
Построив по экспериментальным данным график зависимости ln k от 1/T (т. н. “аррениусовскую зависимость”), можно вычислить энергию активации изучаемой реакции по тангенсу угла наклона, который в этом случае равен -Е*/R (рис. 12.5).
Рис. 12.5. Аррениусовская зависимость и энергия активации |
Дифференцируя логарифмическую форму уравнения Аррениуса по температуре, получим уравнение, подобное уравнению изобары (изохоры) Вант-Гоффа:
d ln k E* ¾¾¾ = ¾¾ dT RT2 |
Интегрирование его в пределах k1 ¸ k2 и Т1 ¸ Т2 приводит к уравнению
k2 E* 1 1 ln ¾ = ¾ (¾ - ¾) k1 R T2 T1 |
или иначе
k2 E* Т2 - Т1 ln ¾ = ¾ ( ¾¾¾) , (12.11) k1 R T1 T2 |
где k1 и k2 - константы скорости данной реакции при температурах T1 и T2 соответственно.
С помощью уравнения (12.11), также называемого уравнением Аррениуса, можно вычислить константу скорости k2 при заданной температуре Т2, если известны значения константы скорости k1 при температуре Т1 и энергия активации реакции Е*. Кроме того, это уравнение позволяет вычислить энергию активации реакции по значениям двух констант скорости при различных температурах:
R T1 T2 k2 E* = ¾¾¾¾ ln ¾ . Т2 - Т1 k1 |
Таким образом, в соответствии с теорией активных соударений повышение температуры увеличивает скорость химических реакций потому, что при этом возрастает доля активных молекул, способных преодолеть потенциальный барьер при столкновении.
12.10. Теория переходного состояния. Активированный комплекс
Расчёты показывают, что для многих химических реакций, если они протекают по механизму непосредственного превращения молекул исходных веществ в продукты, энергии, сообщаемой молекулам при термической активации, недостаточно для преодоления энергетического барьера. Иными словами, при таком механизме энергия активации даже при очень высоких температурах настолько велика, что реакции не должны протекать с заметной скоростью. Тем не менее, химические реакции и в природе, и в промышленных и лабораторных установках идут и часто идут очень быстро. Следовательно, одной теории активных столкновений недостаточно для объяснения причин протекания и механизмов реакций.
В 1930-х г. г. Э. Вигнером, М. Поляни, Г. Эйрингом и М. Эвансом была создана теория, позволяющая объяснить протекание реакций при малых тепловых скоростях молекул. Она носит название теории переходного состояния (или теории абсолютных скоростей реакций). Основные положения этой теории:
1) Взаимодействие молекул не сразу приводит к образованию молекул продуктов. Вначале образуется т. н. “переходное состояние” или активированный комплекс.
2) Активированный комплекс представляет собой неустойчивое образование, в которое входят все атомы столкнувшихся и вступивших во взаимодействие молекул. Время жизни активированного комплекса очень мало; оно измеряется малыми (миллионными, десятимиллионными и т. д.) долями секунды. Расстояния между атомами в активированном комплексе несколько больше, чем в обычных молекулах, поэтому для его образования требуется дополнительная энергия.
3) Энергия активации в связи с этим рассматривается как энергия, необходимая для образования активированного комплекса.
4) Через какое-то время после возникновения активированный комплекс распадается с образованием молекул продуктов; при этом выделяется энергия.
5) Выделяющаяся при распаде активированного комплекса энергия может полностью или частично затрачиваться на активацию других молекул исходных веществ.
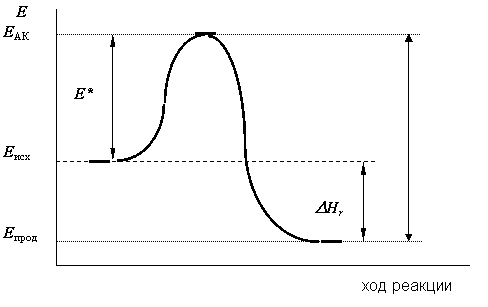
Наглядное представление о протекании реакции во времени в соответствии с теорией переходного состояния может дать энергетический профиль реакции, например, экзотермической (рис. 12.6).
По оси ординат откладывается энергия системы Е, а ось абсцисс - это так называемая координата реакции. Среднему запасу энергии теплового движения молекул исходных веществ соответствует уровень Еисх, энергии, запасаемой в активированном комплексе - уровень ЕАК . Тогда разность ЕАК - Еисх равна величине энергетического барьера, который должны преодолеть молекулы для того, чтобы вступить во взаимодействие энергия активации. Наглядное представление о нём даёт кривая, соединяющая уровни Еисх и ЕАК. Высота энергетического барьера зависит от природы реагирующих веществ, энергии, необходимой для образования активированного комплекса (энергии активации), а также от средней энергии теплового движения молекул Еисх.
Рис. 12.6. Энергетический профиль экзотермической реакции |
При повышении температуры уровень Еисх поднимается, величина энергетического барьера становится меньше и во взаимодействие может вступить большее число молекул. Это и служит причиной ускорения реакции с повышением температуры. При понижении температуры, наоборот, уровень Еисх опускается и величина энергетического барьера возрастает, что приводит к уменьшению скорости реакции.
При распаде активированного комплекса с образованием молекул продуктов выделяется энергия, которой соответствует разность ЕАК - Епрод, где Епрод - средний запас энергии молекул продуктов. Часть этой выделяющейся энергии, равная разности ЕАК - Еисх, пойдёт на активацию новых молекул исходных веществ, а избыток Еисх - Епрод выделится в окружающую среду в виде экзотермического теплового эффекта реакции DНr.
Для эндотермических реакций энергетический профиль выглядит несколько иначе (рис. 12.7). Видно, что в этом случае энергетический уровень Еисх ниже, чем уровень Епрод. В результате этого энергии ЕАК - Епрод, выделяющейся при распаде активированного комплекса, недостаточно для того,
Рис. 12.7. Энергетический профиль эндотермической реакции |
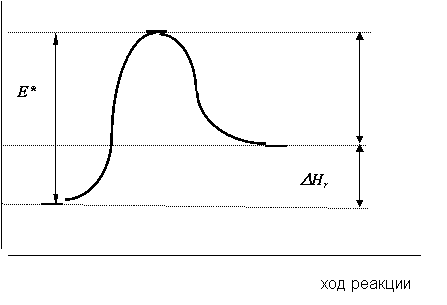
чтобы вызвать активацию новых молекул реагирующих веществ. Поэтому для продолжения реакции необходим подвод энергии извне, в виде эндотермического теплового эффекта.
Существование активированного комплекса подтверждается экспериментальными данными. Так, например, для одной из несложных модельных реакций взаимодействия атома водорода с молекулой водорода
Н2 + Н ® Н + Н2,
значение энергии активации близко к 36,8 кДж/моль. Если бы реакция шла через стадию полной диссоциации молекул Н2, а не через стадию образования активированного комплекса Н2·Н, то потребовалась бы энергия активации 435,1 кДж/моль.
12.11. Гетерогенные реакции
Химические реакции, идущие на поверхностях раздела фаз - гетерогенные реакции - протекают с рядом особенностей, которые связаны, главным образом, с локализацией места взаимодействия между молекулами. К таким реакциям относится очень большое число процессов и, в первую очередь, реакции с участием твёрдых веществ - двух твёрдых веществ друг с другом (т. н. топохимические реакции), твёрдого вещества с газом или с компонентами раствора, в которое оно погружено. Возможны гетерогенные реакции и на поверхностях раздела между двумя жидкими фазами. Поскольку такие лекарственные формы, как, например, смешанные порошки, таблетки, суспензии, эмульсии представляют собой гетерогенные системы, в них возможно протекание гетерогенных реакций указанных типов, что приводит к разложению лекарственных веществ, к изменению физических свойств, к потере потребительных качеств и товарного вида.
Большинство биохимических реакций также идёт на поверхностях раздела, имеющихся в различных структурах организма, т. е. является гетерогенными.
Поэтому выяснение деталей протекания указанных процессов очень важно для биохимии и медицины, в особенности фармакологии. Гетерогенные процессы, происходящие при растворении таблеток, порошков, кристаллизации из растворов имеют большое значение для фармации, в особенности для технологии лекарств.
Гетерогенными являются и электрохимические, и каталитические реакции с участием твёрдых катализаторов. Кроме химических реакций возможны и гетерогенные физико-химические процессы, например, растворение кристаллов и кристаллизация из растворов, адсорбция и десорбция и др.
По сравнению с гомогенными реакциями на общую скорость гетерогенного процесса будут влиять такие дополнительные факторы, как: 1) доставка реагентов к поверхности и 2) удаление продуктов реакции от поверхности. Поэтому большинство гетерогенных процессов включает в себя следующие обязательные стадии:
1) доставка реагентов к поверхности
2) химическое взаимодействие на поверхности
3) удаление продуктов от поверхности
Довольно часто, особенно при реакциях на твёрдых поверхностях, за стадией доставки следует стадия адсорбции молекул реагирующих веществ на поверхности (иногда - с определённой ориентацией), а перед стадией удаления продуктов - стадия десорбции.
1-я и 3-я стадии осуществляются с помощью диффузии реагентов и продуктов, и значит, их скорость будет определяться диффузионными характеристиками как самих реагирующих веществ, так и среды, в которой они находятся. Скорость 2-й стадии будет зависеть от нескольких факторов - от природы веществ, от энергии активации реакции, от концентрации реагентов в приповерхностном слое, каталитических свойств поверхности, иногда от ориентации молекул на твёрдой поверхности и т. д.
Суммарная продолжительность процесса будет определяться скоростью самой медленной (лимитирующей) стадии. Если доставка реагентов и отвод продуктов осуществляются быстрее, чем собственно химическое превращение, то лимитирующей стадией является химическая. В таком случае принято говорить, что гетерогенная реакция идет с химическим (или с кинетическим) контролем. Если же химическое превращение идет быстрее, чем подвод реагентов (или отвод продуктов), то лимитирующей стадией является соответствующий процесс диффузии, и тогда принято говорить о гетерогенном процессе с диффузионным контролем.
ГЛАВА 13
КАТАЛИЗ
13.1. Основные понятия. Значение катализа для медицины,
фармации и биологии
Если энергия активации реакции высока, то лишь небольшая доля сталкивающихся молекул имеет энергию, достаточную для того, чтобы произошла реакция, а если она низка, то реагирует большая часть молекул, и поэтому константа скорости будет большой. Из этого следует, что если каким-либо образом снизить энергию активации, то реакция будет протекать быстрее. И наоборот - при повышении энергии активации реакция будет замедляться. Добиться снижения Е* можно с помощью катализа.
Катализ - это изменение скорости или возбуждение химической реакции веществами (катализаторами), которые участвуют в реакции, но не входят в состав конечных продуктов. Катализатор может многократно участвовать в какой-либо промежуточной стадии химической реакции, и обычно его количество значительно меньше, чем количество реагентов. Реакции в присутствии катализаторов называются каталитическими. Различают положительный катализ (ускорение реакции) и отрицательный катализ или ингибирование (замедление реакции). Однако термин "катализ", как правило, относят к положительному катализу.
Изменение скорости данной реакции при введении в реакционную смесь катализатора характеризуется каталитической активностью. Мерой её может быть увеличение константы скорости, уменьшение энергии активации т. п.
Катализ играет огромную роль в химической промышленности, в том числе при синтезе лекарственных веществ. Многое в природе катализа еще не ясно, но, тем не менее, следует помнить, что различные примеси в лекарственных веществах могут вызвать каталитические реакции их разложения или превращения, что подразумевает необходимость тщательной очистки. Подобную роль могут играть и стенки сосудов, особенно металлических, в которых хранятся лекарственные вещества. Для замедления подобных процессов каталитического разложения, а также фотолиза применяют ингибиторы, антиоксиданты и др.
Огромно значение катализа в процессах, протекающих в живых организмах под действием биологических катализаторов - ферментов (ферментативный катализ).
13.2. Краткий исторический очерк
Природными катализаторами - ферментами при изготовлении алкогольных напитков и уксуса, сыроварении, хлебопечении, выделке кож и т. д. люди пользовались еще в глубокой древности.
Первые научные сообщения и катализаторах неферментной природы появляются в ХVIII веке. Так, было описано каталитическое ускорение реакций этерификации (К. В.Шееле, 1782), дегидратации (Дж. Пристли, 1778), гидролиза полисахаридов (, 1811).
Термин "катализ" был предложен в 1835 г. . В учении о катализе большой вклад был внесён русскими химиками (1871 - открытие реакции гидратации ацетилена в присутствии серной кислоты и ртутных солей), (1877- восстановление различных органических соединений над платиной), шуткиным (1877 - реакции этерификации), , .
Учение о катализе нашло дальнейшее развитие в исследованиях таких видных ученых, как П. Сабатье, , и др.
13.3. Виды катализа
Существует такая классификация видов катализа:
- По агрегатному состоянию принято различать гомогенный катализ, при котором реагирующие вещества, продукты реакции и катализатор находятся в одной фазе, обычно жидкой или газовой, и гетерогенный катализ, при котором реагирующие вещества и продукты находятся в одной фазе, обычно жидкой или газообразной, а катализатор - в другой, обычно в твёрдой. Существует также гетерогенно-гомогенный катализ, при котором реакция, начинаясь на поверхности катализатора, продолжается в объёме жидкой или газовой фазы.
- По химической природе катализатора различают кислотно-основный катализ, при котором реакции протекают в присутствии кислот или оснований, катализ на металлах, катализ на оксидах и т. д. Особую группу каталитических процессов составляют реакции ферментативного катализа. Для некоторых реакций катализатором может служить их собственный продукт. Такие реакции называются автокаталитическими.
- По избирательности действия. Во многих случаях один и тот же катализатор может изменять скорость нескольких возможных между данными реагирующими веществами реакций (неспецифический катализ), но существует и широко применяется на практике избирательный (или специфический) катализ, когда катализатор из многих возможных в данной реакционной смеси реакций "выбирает" одну. Это используется, например, для синтеза стереорегулярных полимеров. Высочайшей избирательностью обладают биологические катализаторы - ферменты.
- По физическому состоянию твёрдого катализатора различают катализ на компактных металлах, когда катализатор представлен каким-либо одним металлом в различной степени раздробленности (дисперсности); катализ на носителях, когда катализатор-металл напыляется тонким слоем на какой-либо инертный носитель (фарфор, асбест, активированный уголь); катализ на смешанных катализаторах, когда катализатор представляет собой смесь порошков различных металлов, оксидов или других веществ.
13.4. Механизм действия катализаторов
Единой теории катализа до настоящего времени нет. Однако накопленный экспериментальный материал позволяет сделать заключение о том, что катализатор способен входить в состав промежуточного соединения (активированного комплекса). При этом снижается энергия активации и происходит ускорение как прямой, так и обратной реакции. Важно понимать, что катализатор не может вызвать термодинамически невозможный процесс. Более того, катализатор не смещает химического равновесия, а лишь ускоряет его достижение.
О том, что катализатор участвует в химической реакции, говорит, например, то, что физическое состояние катализатора в ходе реакции может существенно изменяться. Так, крупнокристаллический диоксид марганца, катализирующий распад бертолетовой соли, после реакции превращается в мелкий порошок. В принципе это изменение степени дисперсности может отразиться на каталитической активности, но в небольшой степени.
Поэтому под неизменяемостью катализатора в ходе реакции имеют в виду постоянство его количества и химического состава до и после реакции.
13.5. Гомогенный катализ
Многие реакции в газовой фазе носят отчётливо выраженный каталитический характер. Таковы, в частности, процессы, катализатором которых является молекулярный иод. Например, скорость реакции разложения диэтилового эфира
C2H5OC2H5 ® C2H6 + CH4 + CO
в присутствии иода увеличивается по сравнению с некаталитическим пиролизом примерно враз. В данном случае это связано с изменением механизма реакции. Первичный процесс заключается в расщеплении молекулы эфира под действием иода и в образовании в качестве промежуточных соединений иодэтана, иодида водорода и уксусного альдегида:
C2H5OC2H5 + J2 ® CH3CH2J + HJ + CH3CHO
Вторичные процессы, приводящие к образованию продуктов реакции, заключаются в образовании этана с регенерацией катализатора:
CH3CH2J + HJ ® C2H6 + J2
и в распаде уксусного альдегида:
CH3CHO ® CH4 + CO
По сравнению с некаталитической реакцией (Е* = 216,73 кДж/моль) в этой каталитической реакции энергия активации снижается до 74,475 кДж/моль.
Гомогенно-каталитические реакции особенно распространены при проведении процессов в жидкой фазе. В большинстве случаев при этом приходится встречаться с кислотно-основным катализом. К таким процессам относятся ускоряющиеся под действием водородных ионов реакции этерификации и гидролиза сложных эфиров, инверсии сахарозы и т. п. (кислотный катализ). И в этих реакциях роль катализатора (ионов Н+) формально сводится к снижению энергии активации. В некоторых случаях кислотно-основного катализа катализатором являются ионы ОН-, иногда недиссоциированные молекулы слабых кислот и оснований. В общем, можно сказать, что кислотно-основный катализ основан на протолитическом взаимодействии между катализатором и реагирующим веществом, как правило, не обладающим ярко выраженными кислотными или основными свойствами. Для реакций, протекающих по механизму кислотно-основного катализа, характерно то, что они не сопровождаются разрывом электронных пар в реагирующих веществах. К гомогенному катализу можно отнести и переваривание жидкой пищи в желудке под действием желудочного сока.
С. Аррениусом было установлено, что каталитическое действие кислот может быть усилено добавлением в реакционную смесь нейтральных солей, не имеющих общих анионов с кислотой (первичный солевой эффект), или имеющих такие общие анионы (вторичный солевой эффект). Природа солевых эффектов пока неясна.
13.6. Гетерогенный катализ
На практике наиболее часто встречаются два типа гетерогенного катализа, при которых катализатор находится в твёрдой фазе: 1) процессы с реагирующими веществами в жидкой фазе; 2) процессы с реагирующими веществами в газовой фазе.
Твёрдые катализаторы при этом могут иметь самую различную химическую природу (металлы, оксиды и др.)
Реакция, как правило, происходит на границе раздела фаз, то есть на поверхности катализатора. Как и в случае других гетерогенных реакций, каталитический процесс состоит из нескольких стадий. Но, поскольку процессы адсорбции в гетерогенном катализе играют чрезвычайно важную роль, таких стадий имеется как минимум, пять: 1) подвод (диффузия) реагирующих веществ к поверхности катализатора; 2) адсорбция их молекул (иногда с определённой ориентацией); 3) реакция на поверхности катализатора; 4) десорбция продуктов; 5) отвод (диффузия) продуктов реакции от поверхности. Как правило, действие катализатора проявляется на 2-й, 3-й (в особенности) и на 4-й стадиях.
Важным свойством твёрдых катализаторов является уже упоминавшаяся избирательность (специфичность) действия, под которой понимается способность ускорять одну из нескольких возможных в данной реакционной смеси реакций. Например, из смеси СО с Н2 ("водяной газ") в зависимости от природы катализатора и от условий могут получаться различные продукты:
1) CO + 3H2 ¾¾¾¾¾¾¾¾® СH4 + H2O
Ni; Т = oC
2) CO + 2H2 ¾¾¾¾¾¾¾¾¾¾® CH3OH
Cu; повышенное давление
3) CO + H2 ¾® смесь олефинов и парафинов + Н2О
Co
Причины избирательного действия катализаторов могут быть различными, причём многие из них пока не выяснены. Часто для придания большей избирательности, повышения термической стойкости и механической прочности, а также для повышения активности катализаторы применяют не в виде чистых веществ, а в виде сложных многокомпонентных систем. Наиболее интересны три типа таких катализаторов: смешанные, на носителях и промотированные.
Смешанные катализаторы, как правило, представляют собой смесь двух или нескольких оксидов, например Al2O3 + ThO2 ; Al2O3 + Cr2O3 ; CoO + MgO. Состав этих катализаторов можно изменять, и активность их часто зависит от состава.
Катализаторы на носителях представляют собой какое-либо инертное по отношению к данной реакции вещество, на которое нанесён различными способами (напылением, осаждением из раствора, пропиткой раствором соли с последующим восстановлением металла и т. п.) катализатор. Обычно катализ на носителях применяется с целью удешевления производственного процесса, так как в качестве катализаторов часто используются достаточно дорогие металлы - никель, кобальт, платина, золото и др.
Промотированные катализаторы - катализаторы с добавлением промотóров, то есть веществ, которые сами по себе не обладают каталитическими свойствами по отношению к данной реакции, но увеличивают активность используемого катализатора.
Некоторые добавки могут вызвать не промотирование, а обратный эффект - "отравление" катализатора, то есть снижение его каталитической активности. Такие добавки называются каталитическими ядами. Их действие объясняется хемосорбцией на поверхности катализатора или другим блокирующим механизмом.
Необходимо помнить, что разложение лекарственных веществ – тоже химическая реакция, которая может катализироваться различными твёрдыми веществами, находящимися с ними в контакте. В качестве таких нежелательных катализаторов могут выступать неудалённые при очистке примеси непрореагировавших исходных веществ или побочных продуктов, сопутствующие вещества, а в особенности - материалы упаковки, главным образом, металлы. Поэтому для продления срока годности лекарств немаловажными факторами являются тщательная очистка их от примесей и подбор неактивного в каталитическом отношении материала упаковки.
13.7. Теории гетерогенного катализа
По мере накопления опытных данных о гетерогенных каталитических реакциях неизбежно встает вопрос о природе и строении активной поверхности катализатора.
Тот факт, что для отравления катализатора вполне достаточно ничтожно малого количества каталитического яда, а также что каталитическая активность практически не изменяется при напылении катализатора на носитель (где он присутствует в виде очень небольших частичек, часто коллоидных размеров), говорит о существовании на поверхности катализатора каких-то активных центров. Эти активные центры, на которых собственно и идет каталитический процесс, должны занимать, по-видимому, очень малую часть общей поверхности катализатора, а остальная часть её при этом остается инертной. Природа активных центров может быть различной (выступы на поверхности, определённые участки кристаллической структуры и т. д.), но несомненно, что она играет весьма существенную роль в процессе каталитического превращения.
Различные теории катализа по-разному интерпретируют понятие активного центра и роль этих центров в каталитическом действии.
Рассмотрим некоторые из них.
13.7.1. Мультиплетная теория
В 1929 году впервые были высказаны положения мультиплетной теории:
1) Активный центр представляет собой совокупность определённого числа адсорбционных центров, расположенных на поверхности катализатора в геометрическом соответствии со строением молекулы, претерпевающей превращение (принцип геометрического или структурного соответствия). Согласно теории Баландина, таким активным каталитическим центром является участок поверхности катализатора с определённой кристаллической структурой, на котором расстояния между центрами атомов соответствуют расстояниям в адсорбирующихся и превращающихся молекулах. Активный центр, состоящий из двух атомов, был назван дублетом, из трёх - триплетом, из четырёх - квадруплетом, из шести атомов - секстетом. Общее название активного центра, состоящего из нескольких атомов - мультиплет.
2) При адсорбции реагирующих молекул на активном центре образуется мультиплетный комплекс, в результате чего происходит перераспределение связей, приводящее к образованию продуктов реакции.
Таким образом, мультиплетная теория ставит геометрическую структуру реагирующих на поверхности катализатора молекул в прямую зависимость от геометрического расположения адсорбционных центров. Это может объяснить причины специфичности каталитического действия. Например, реакция дегидрирования циклопарафинов
|
|
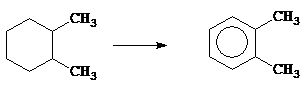
катализируется металлами, кристаллизующимися в кубических гранецентрированных или гексагональных решётках, так как только на октаэдрических гранях первой и на базопинакоидах второй встречается расположение атомов решётки, соответствующее строению шестичленных циклов. Однако из таких металлов катализаторами являются только те, у которых расстояние между центрами атомов в решетке лежит в пределах от 2,77´10-8 см (Pt) до 2,48´10-8 см (Ni). Так, каталитически активны Рd (2,74´10-8 см), Ir (2,70´10-8 см), Rh (2,68´10-8 см), Сu (2,56´10-8 см), а кристаллографически аналогично построенные Тh (3,60´10-8 см), Аg и Аu (2,88´10-8 см) при дегидрировании шестичленных колец каталитически неактивны.
В 1935 году включил в мультиплетную теорию принцип энергетического соответствия, согласно которому энергия активации катализируемой реакции зависит и от энергии связи между катализатором и мультиплетным активированным комплексом (а не только от энергии связи между составными частями реагирующих молекул). Отсюда следует, что чем больше сродство катализатора к адсорбирующимся исходным веществам, тем заметнее снижается энергия активации катализируемой реакции.
13.7.2. Теория активных ансамблей
Эта теория была создана (1939). Она наиболее подходит для описания катализа на носителях.
Кобозеву поверхность носителя (активированный уголь, асбест, пемза и т. п.) имеет блочное строение, то есть состоит из отдельных участков - блоков, которые отделены друг от друга дефектами поверхности, например, микротрещинами, включениями примесей и т. п. Эти поверхностные блоки представляют собой области свободной миграции атомов нанесённого металла, способных перемещаться по их поверхности в результате теплового движения. При попадании на такой блок определённого числа атомов (зависящего от условий нанесения), они в результате миграции способны образовать активный центр, названный активным ансамблем, на котором и происходит каталитическое превращение. Активный ансамбль состоит из нескольких атомов, являющихся как бы двухмерным зародышем кристаллической решётки. Если его геометрическое строение соответствует строению молекул реагирующих веществ, то они способны адсорбироваться на нём и вступать в каталитическую реакцию. Другие положения теории в основном совпадают с положениями мультиплетной теории.
13.7.3. Электронная теория
В основе электронной теории, разработанной и Ф. Ф.Волькенштейном (1940), лежит положение о том, что на поверхности катализатора имеются нескомпенсированные валентности, обусловленные свободными или слабосвязанными электронами, присутствующими в его кристаллической структуре. Адсорбция реагирующих молекул происходит с участием этих нескомпенсированных валентностей катализатора, благодаря чему связи внутри молекул ослабляются настолько, что они, по сути дела, представляют уже свободные атомы или радикалы. Продукты реакции образуются при взаимодействии этих атомов или радикалов, находящихся в непосредственной близости друг к другу. Эта теория довольно хорошо описывает катализ на переходных металлах, у которых имеется незавершённая d-электронная оболочка.
13.8. Ингибиторы
Ингибиторы - это вещества, замедляющие протекание реакций. Как и катализаторы, они обладают специфичностью действия.
Возможны такие механизмы действия ингибиторов:
1) взаимодействие с промежуточными продуктами с образованием соединений, малоактивных в последующих реакциях (поэтому ингибиторы особенно эффективны в случае цепных реакций);
2) взаимодействие с активными центрами катализаторов (отравление);
3) блокирование активных центров катализаторов (когда, не занимая активных центров, ингибиторы препятствуют подходу к поверхности катализатора реагирующих веществ).
Во многих случаях использование ингибиторов приносит большую пользу, например, при защите металлов от коррозии, при продлении сроков годности лекарственных препаратов и пищевых продуктов с использованием ингибиторов-антиоксидантов. На ингибировании различных внутриклеточных реакций болезнетворных микроорганизмов, приводящем к угнетению их роста или размножения, основано фармакологическое действие некоторых лекарственных веществ, например, сульфаниламидов.
В других случаях ингибирование реакций может иметь отрицательное значение. Так, действие многих биологических ядов связано с ингибированием ферментативных реакций в организме. Случайное попадание веществ, являющихся ингибиторами, в реакционные аппараты приводит к замедлению химических реакций, а часто и к полному отравлению катализатора, что требует его замены.
VI. Ф О Т О Х И М И Я
ГЛАВА 14
ФОТОХИМИЧЕСКИЕ РЕАКЦИИ
Кроме рассмотренной термической активации возможна и другая активация молекул - фотохимическая. При термической активации атомы и молекулы участвуют в реакции в их основных электронных состояниях. Фотохимические реакции осуществляются лишь после перевода молекул исходных веществ в электронно-возбуждённое состояние под воздействием электромагнитного излучения. Таким образом, фотохимические реакции - это реакции, происходящие только при наличии электромагнитного излучения в широком интервале энергий - от видимого и ультрафиолетового света до рентгеновских и g-лучей.
Таким образом, фотохимия это - раздел химической кинетики, занимающийся поведением электронно-возбужденных молекул. В зависимости от системы и от условий, в которых происходит фотовозбуждение, такая молекула может участвовать в нескольких различных процессах. Она может потерять свою избыточную энергию при соударении с другой молекулой без химического взаимодействия (при этом выделяется теплота); возбужденная молекула может вернуться в основное состояние, испустив фотон, т. е. путём флуоресценции или фосфоресценции. Наконец, она может претерпеть химическое превращение, например, изомеризацию, ионизацию, диссоциацию на атомы или радикалы.

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 |
